

CRAやCRCさんが苦労をして集めた治験のデータは治験総括報告書にまとめられることになりますが、実は中身をじっくり見たことが無いという人も多いのではないでしょうか?
そこで今回は、治験総括報告書の概要とCRA/CRCさんとして注目すべきポイントをご紹介していきます。
治験総括報告書の概要

ではでは、「そもそも治験総括報告書って何だっけ…?」というところからお話をしていきましょう。
治験総括報告書については、GCP第25条に規定されています。
第25条
治験依頼者は、治験を終了し、又は中止したときは、総括報告書(治験の結果等を取りまとめた文書をいう。以下同じ。)を作成しなければならない。
以上。
分かりましたか?
GCPじゃこれだけなので、正直よく分からないですよね。GCPの座学でもやったかもしれませんが、「治験が終わった時に出す資料」と暗記をして終了ですね(はい、CRAの頃の私はそんな感じでした…)。
座学は半分眠って真面目に受けていましたが、正直今一ピンとこないままCRAをやっていたような気もします。
そのおかげで、振り返ってみれば本質をあまり理解しないまま仕事をしてしまっていたなと思うことも多々あったので、今回ご紹介をしようと思い立ちました。
モニターの人、総括報告書を読んだことが
— りょう@CRA (@CRA92475462) March 25, 2023
きっかけをありがとうございました!
ちなみに、1点だけ注意をするとしたら治験総括報告書のフルバージョンは通常外部には出さないという点です。
ネット上で閲覧できるのは大抵「治験総括報告書の“概要”」であり、透明性確保のために外部向けに調整をした資料になります。
ですので、有効性や安全性にフォーカスした内容の提示になるので「これがフルバージョンではない」ということは念頭に置いておきましょう(概要には大抵逸脱などの情報は記載されていない)。
ちなみに余談ですが、業務上では「治験総括報告書」と呼ぶと長いので大体の方は「CSR」と呼んでいます。
プロっぽくいきたい感じであれば、「あぁ、CSRね、うんうん」という感じに使ってみるとそれらしくなりますよ(フフフ
治験総括報告書(CSR)の位置付け
医薬品の承認申請をするためには規制当局(PMDA)にコモン・テクニカル・ドキュメント(Common Technical Document)を提出することが義務付けられています。
昔は、各国それぞれの規制で決められた提出資料を出していて国ごとにバラバラだったのですが、ルールをハーモナイズすることによって色々な国での承認を迅速化させようとICHがCTDのガイドラインを出しました。
構成はこんな感じです。
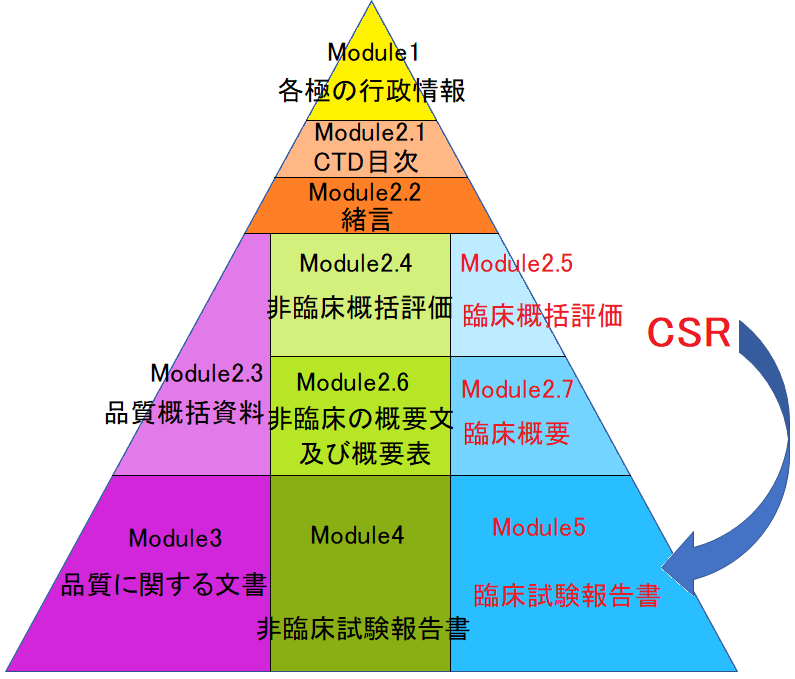
この中のModule 2.5(臨床概括評価)とModule 2.7(臨床概要)には治験の内容が記載されるのですが、ここの根拠資料として治験総括報告書(CSR)が添付されます。
「本剤はプラセボと比較し、主要評価項目である●●の数値が有意に高かった。」とCTDに記載した場合、「その根拠は?」となる訳です。
そこで治験総括報告書(CSR)が添付されているということですね。
今回の記事では、ICHをまだほとんど読んだことがなく、どこから手を付けたら良いか分からない方向けに、簡単な概要をまとめてみました。 また、2021年7月7日(水)に第43回ICH即時報告会がありましたので、その内容についても簡単に触れています。 “ICHは難しそう”と苦手意識を持っている方や新人の方、またこれから臨床開発の世界で働く予定の方の一助となれば幸いです!

治験総括報告書(CSR)に求められていること
治験総括報告書(そろそろ慣れてきたと思うので、以下「CSR」にしますね)は、当局が治験の結果を確認するために使われます。
そして、当局としてどういうことを記載して欲しいかは「治験の総括報告書の構成と内容に関するガイドラインについて」にバッチリと書かれています。
ですが…
ここで依頼者がイケていないと問題が起こるんです。何だと思いますか?
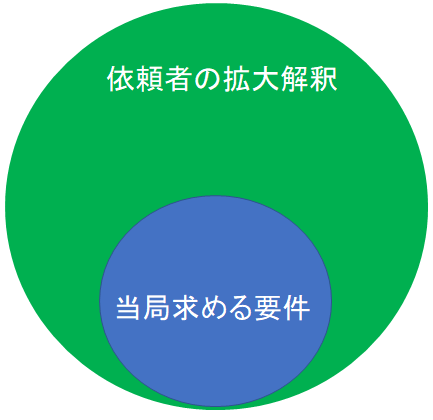
当局がガイドラインで示している要求事項を依頼者が拡大解釈してしまうと、オーバークオリティのCSRが出来上がってしまうのですよ。
製薬メーカー「当局はきっとここまで求めてくるだろう。なので、ここまでCSRに書いておきたい。」
私たち(少なくとも弊社)は、治験を始める際には大まかにどのようなストーリーでCSRを書くかを決めます。
「添付文書→CTD→CSR→PRT→手順書」というようにゴールから逆算して決めていきますが、上流でオーバークオリティが発生したらどうなるか?
はい、オーバークオリティのPRTや手順書が出来上がってCRAやCRCさんが悲鳴をあげることになります。
だから上流でオーバークオリティを食い止めなければいけない。
なので、私はCRA経験者として色々口を出すわけですね。 上流で決まってしまったらPRTや手順書はそれに沿って作るので上流で食い止めようというわけです。上流で食い止めないとCRAやCRCさんが激流に流されちゃいますし(上手いこと言った!)。
話が反れちゃいましたが、私が言いたかったのはCSRの要求事項は適切に汲み取って欲しい、そして、CRAからMedical Writing(MW)にキャリアチェンジをしようと思う方には是非ここを武器にして欲しいとも期待しています(>私にご相談いただいた数名の方々!応援しています!)。
CSRには監査証明書を添付する
治験で監査を実施した場合には、監査での指摘事項を報告する「監査報告書」と、監査をやったよという「監査証明書」があります。
新人の頃は混同しやすいですよね。ぺこさんのツイートのアンケートからもその混同っぷりが分かるかと思います。
総括報告書と一緒に保管しておくものは監査報告書である
— ぺこ (@peko_peko9) October 27, 2020
これはですね、当局の気持ちになって考えてみると良いと思います。
当局から見てCSRというのは、「治験の結果を確認する資料」でしたね。
参照資料の信頼性が担保されていないとその資料を参照するのはNGですよね?なので、当局はしっかりと信頼性が担保されていることを“確認”したい。
だから、「監査証明書」を添付させてその資料の信頼性を保証(証明)して欲しいのです。
事実を確認した上で判断をするのは当局なので、監査報告書はいらない(ということだと私は思っています)。
例えば監査で「これはGCP不遵守」と言われていたとしても当局が問題無いと判断すれば問題無いということでしょうし(ほぼそのパターンは無いと思いますが…)、逆に監査で「問題がありませんでした」となっても、当局がGCP的に問題だと判断すれば問題になりますからね。
CSRでCRA/CRCさんが見ておくと勉強になるポイント

項目のフルバージョンは「治験の総括報告書の構成と内容に関するガイドラインについて」を参照いただきたいのですが、CSRの中でCRA/CRCさんが確認しておいても良いかなと思うのは以下の項目になります。
11.1 解析したデータセット
11.4 有効性に関する成績及び個別患者データ一覧表
11.4.2.2 脱落又は欠測値の取扱い
12.2.1 有害事象の簡潔な要約
12.2.2 有害事象の表示
12.2.3 有害事象の分析
12.3.2 死亡,その他の重篤な有害事象及び他のいくつかの重要な有害事象の叙述
12.3.3 死亡,その他の重篤な有害事象及び他の重要な有害事象の分析及び考察
12.4.2 各臨床検査項目の評価
12.6 安全性の結論
13 考察と全般的結論
ちなみに、テンプレートの1章~9章までの内容はプロトコルの内容とほぼ同じなのであまり見なくてもOKです。
あとは、CSRの章は必ずテンプレート通りでなくてはいけないということではないので、CSRによっては章番号がずれている点をご留意下さい。
ざっくりとですが、着目ポイントを見ていきましょう。
10章 治験対象患者
10章の10.2 「治験実施計画書からの逸脱」の項目では、重要な逸脱ついて以下のように分類して施設毎に逸脱の件数がまとめられています。
- 組み入れ基準を満たしていないにもかかわらず,治験に組み入れられた患者
- 治験期間中に中止基準に該当するようになったが,中止されなかった患者
- 治療方法や用量が不適切であった患者
- 禁止されている併用療法を受けた患者
治験総括報告書の概要には10章の内容が掲載されていないことがほとんどなので、あまり実際の記載を見る機会がないかもしれません。
また、上記の項目はガイドラインに示されたものですので、上記+αの項目を設定している依頼者もいると思います。
イメージとしてはこのような感じです。
| Noris-123群 (n=50) n(%) |
Placebo群 (n=50) n(%) |
Total (n=100) n(%) |
|
| 組み入れ基準を満たしていないにもかかわらず,治験に組み入れられた患者 | 1 (2.0) |
0 | 1 (1.0) |
| 治験期間中に中止基準に該当するようになったが,中止されなかった患者 | 0 | 0 | 0 |
| 治療方法や用量が不適切であった患者 | 0 | 0 | 0 |
| 禁止されている併用療法を受けた患者 | 2 (4.0) |
1 (2.0) |
3 (3.0) |
| 主要評価項目に影響を及ぼすその他の逸脱がある患者 | 0 | 1 (2.0) |
1 (1.0) |
| Noris-123群 (n=50) n(%) |
Placebo群 (n=50) n(%) |
Total (n=100) n(%) |
|
| 組み入れ基準を満たしていないにもかかわらず,治験に組み入れられた患者 | 1 (2.0) |
0 | 1 (1.0) |
| 治験期間中に中止基準に該当するようになったが,中止されなかった患者 | 0 | 0 | 0 |
| 治療方法や用量が不適切であった患者 | 0 | 0 | 0 |
| 禁止されている併用療法を受けた患者 | 2 (4.0) |
1 (2.0) |
3 (3.0) |
| 主要評価項目に影響を及ぼすその他の逸脱がある患者 | 0 | 1 (2.0) |
1 (1.0) |
例えば、ここで当局が求めていること以上のことを設定すると、それがプロトコルに反映されて…あとはお分かりですね?
11章 有効性の評価
11章の11.1 「解析したデータセット」では、有効性の解析から除外された患者とその理由についてが記載されています。
イメージとしては以下のような感じです。

この試験では、有効性評価の許容範囲にデータが存在しないという理由でFASから除外されている症例が1例いることが分かりますね。
当然のことながら、せっかく治験に参加して下さった被験者さんのデータが解析に使われないことはなんとか避けたいものです。
解析から除外されてしまうというパターンはそれほど多くはないものの、どのような状況になってしまうと解析から除外されてしまうのかという事例を蓄積しておくことで担当のプロジェクトでの発生を防げる可能性があります。
どのような場合に解析から除外されるかは、治験実施計画書(PRT)や統計解析計画書(SAP)に記載されています。
自分の担当試験では解析除外とならないよう事前の対策を取っておくことやCSRを確認できる機会があればどのような状況で解析から除外されてしまったのかを知っておくと今後の引き出しが増えるかと思います。
12章 安全性の評価
12.3.2 「死亡,その他の重篤な有害事象及び他のいくつかの重要な有害事象の叙述」などでは、個々の死亡や重篤な有害事象について簡潔な叙述を記載するよう求められています。
そして、叙述には以下のことを記載するようガイドラインでは定められています。
- 事象の種類と強さ,事象発現までの臨床経過,治験薬の投与に関連する発現時期;関連する臨床検査値,投薬が中止されたかどうか及び中止の時期;対策としてとられた処置,死後所見,因果関係についての治験責任医師の意見及び適切であれば因果関係についての治験依頼者の意見
- 患者識別コード
- 患者の年齢及び性別;適切であれば患者の全身の臨床的状態
- 治療がなされている疾患(全ての患者が同じ疾患であれば必要ない)及び罹病期間(現在のエピソードの期間)
- 関連する合併症・既往症及びその発症・罹病期間の詳細
- 関連する併用薬・前治療薬及びその用量の詳細
- 投与された治験薬名,患者間で薬剤の用量が一定でなければその用量及び投与期間
CSR内で個々の症例レベルで叙述が要求されているのは、死亡・その他の重篤な有害事象と重要な有害事象についてです。つまり、SAE報は上記の叙述で記載すべき内容がしっかりと読み取れる内容を作成すること/モニタリングすることが重要なのです。
その他のAEについては叙述が求められていないことからも分かる通り、あまりに詳細な情報は求められていません。
もちろん、EDCの収集項目として設定されている部分に関してはデータの質を担保するためその根拠をしっかりと確認しておく必要がありますが、それ以上の情報はオーバークオリティです。
時々、必要以上の情報をMVR(モニタリング報告書)に記載するよう指示する依頼者がいますが、実はCSRのガイドラインの観点からはズレている可能性があったりします。
さて、叙述が実際どのような感じで書かれているか見てみましょうか。
一般的にオープンにされている治験総括報告書の概要はこんな感じです。
 出典:ASP8825 長期投与試験 治験総括報告書 概要
出典:ASP8825 長期投与試験 治験総括報告書 概要
あらら、これじゃ分かりませんね。みなさんにイメージを持ってもらうため特別にサンプルを書いてみようと思います。
被験者NRS-01-001(男性、80歳)はNoris-123群に組み入れられ、2023年1月1日にNoris-123の投与を開始した。
被験者には2023年1月11日(Day 11)にCTCAE Grade2の肺炎が発現し、2023年1月29日(Day 29)に消失した。
経過として2023年1月11日(Day 11)の朝より咽頭痛と38℃の発熱があり外来を受診し、CRP及びPCT高値を認めたため、即日入院となった。2023年1月15日にCRPの低下と解熱傾向を認め、2023年1月29日にCRP及び発熱が回復したため退院となった。
肺炎の処置としてソリューゲンF注 500mLを1月11日から1月12日まで、メロペネム3gを1月11日から1月16日まで、ソルデム3A輸液500mL及び塩化ナトリウム500mLを1月12日から1月16日まで、スルバクタムナトリウム・アンピシリンナトリウム 6gを1月16日から1月26日までそれぞれ静注した。
本事象は、発熱性好中球減少症に続発する事象であると考えられ、治験責任医師により治験使用薬との因果関係は否定できると判断された。
叙述の書き方は色々あると思います。
今回は全て文章で書いてみましたが、実際には表でまとめて書かれていることもあったりします。
大事なのは、発生したSAEに対して時系列に沿って適切に情報が集められているかという点ですので、モニタリングをする際も流れをイメージしながらおこなうことで、CSRに求められる要件もクリアできることになります。
CRAやCRCさんであれば必ず直面する有害事象の因果関係判定の確認。 これがまた厄介で、依頼者や先輩によって考え方に差があることもあり、特に新人さんは何が正しいのか混乱してしまう部分かと思います。 今回は根拠資料をしっか …

13章 考察と全般的結論
13章では、有効性と安全性について簡潔に要約することが求められています。
詳細は以下のような感じです。
有効性と安全性の結果及びリスク・ベネフィットとの関係を必要に応じて表,図及びこれまでの章で述べてきたことを引用して簡潔に要約し,考察すること。
その要約は,単に結果の記述を繰り返したり,新しい結果を紹介するものであってはならない。
考察及び結論では,全ての新しい又は予想外の所見を明確にし,その意義について説明すること。さらに,関連する測定値間の不一致など,可能性のある全ての問題について論じること。また,他の既存のデータを考慮し,結果の臨床的適切性及び重要性についても論じること。
個々の患者又はリスクグループに対する個別の有益性又は特に必要とされる注意事項,及び今後の治験実施のためのあらゆる意味合いを明らかにすること。あるいはそのような考察は,申請資料全体を参照している安全性,有効性の要約(integrated summaries)に示してもよい。
この章では、治験で頑張って集めてきた有効性と安全性のデータから治験の結果が考察され依頼者見解が述べられる部分です。
これを見て何か日々の業務に活かす…というものではないのですが、治験のデータが最終的にどのように要約されてまとめられるのかは見ておいても良いのかなと感じたので入れてみました。
そして、一般公開されている「治験総括報告書の概要」にはこの13章の結果を載せていることが多いので(更に要約されている場合もあり)、この部分はみなさんでもネットで検索をすれば見つけることができます!
有効性の部分は以下のような感じです。
ASP8825のレストレスレッグス症候群に対する有効性が記載されていますね。
 出典:ASP8825 長期投与試験 治験総括報告書 概要
出典:ASP8825 長期投与試験 治験総括報告書 概要
まとめ
今回は治験総括報告書について、概要と着目ポイントについてまとめてみました。
治験総括報告書は薬事承認申請で提出するCTDの添付資料の1つという位置付けで、治験の結果が事実として記載されている資料です。
治験総括報告書では事実を淡々と記載して、製薬企業としての主張はCTD(M2.5)でやっていくことになります。製薬企業が当局に対して想いを主張できる場ということですね。
日々の業務がどのような形で報告書にまとめられているかを知ることでどこに意識をして仕事をすれば良いのか考えるきっかけになれば嬉しいです。
依頼者の立場でこの記事を読んでいる方は、オーバークオリティに注意していきましょう!私も今後とも気を付けていきたいと思います!




.jpg)

“【CRA/CRCも知っておきたい】治験総括報告書の確認ポイントを分かりやすく解説!” への1件のフィードバック