

今回の記事では、ICHをまだほとんど読んだことがなく、どこから手を付けたら良いか分からない方向けに、簡単な概要をまとめてみました。
また、2021年7月7日(水)に第43回ICH即時報告会がありましたので、その内容についても簡単に触れています。
“ICHは難しそう”と苦手意識を持っている方や新人の方、またこれから臨床開発の世界で働く予定の方の一助となれば幸いです!
はじめに

業界で働かれている方は、ICHの重要性はよくご存じのことかと思いますが、本ブログを読んで下さっている方の中には多くの初学者の方、そして就活生の方もいます。
私がCRAとして働き始めて間もない頃は、ICHに関しては取っ掛かりにくく、重要なものとは分かりつつも内容の理解を後回しにしてしまっていました。
「なんだかすごく難しそうだな」と漠然と考えていたのですよね。
なので今回の記事では、初学者の方でもなるべく取っ掛かりやすいように、あえて基礎的な説明や簡単な言葉を使いながらご紹介をしていきたいと思います。
熟練の業界の皆様にとってはもどかしい部分もあるかと思いますが、お付き合いいただけますと幸いです。
ICHとは?
![]()
ICHとは、International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Useの略で、医薬品規制調和国際会議のことです。
かつて、日・米・欧では、新薬の薬事承認には、品質・有効性・安全性のデータを報告し評価することを求めるという大枠の認識は共通であったものの、細かい部分を見てみると、求められる要件が異なっているという問題がありました。
その為、製薬会社各社は、日・米・欧全ての国で承認を得ようとすると、重複した治験を行う必要があり、コスト面、時間面において非常に非効率的な状況でした。
それではいけないと、日・米・欧の各医薬品規制当局と業界団体の6団体が1990年4月に発足させたのがICHになります。
現在では、国際共同治験も増えてきており、世界的に目線を合わせることの重要性が一層増している状況と言えます。
ICHでは、上記でお話したような手続き上の都合で目線を合わせるだけではなく、より高品質な医薬品開発や倫理的な側面からの世界的調和も目的としていることが特徴です。
ICHガイドラインの種類
ICHでは、調和を図るために各ガイドラインを作成しているのですが、ガイドラインを読み進める上では、QSEMの意味も頭に入れておくと理解が進みます!
●Safety(安全性)
●Efficacy(有効性)
●Multidisciplinary(複合領域)
ガイドラインでは、それぞれの頭文字+数字で構成されています。
例えば、このあとご紹介するE6とE8はそれぞれ頭文字に「E」とあるので、Efficacy(有効性)に関連したガイドラインということが分かりますね。
この中で、Mだけ少し分かりにくいのですが、これは、QSEの3領域に共通する内容を取り扱っているガイドラインで、例えば、M12「薬物間相互作用」やM13「即放性経口固形製剤の生物学的同等性試験」などがあります。
CRCさんやCRAは、少なくともEに分類されるICHに関わりが深いことが多いので、しっかりとチェックしておきたいところです。
ガイドラインの作成段階
それぞれのガイドラインは、以下のようなステップを経て完成します。
ガイドライン(GL)を作成するトピックが総会で承認を受けると、専門家作業部会(Expert Working Group:EWG)が設置される。その後、EWGで技術ドキュメント(GL案のベース)が作成される。
【Step2a】
EWGで作成した技術ドキュメントが総会で承認される。
【Step2b】
技術ドキュメントをベースに作成したGL案が総会の規制当局代表者によって承認される。
【Step3】
ICHの各地域・国の規制当局(日本では厚生労働省)からガイドライン案が公表され、公に意見(パブリックコメント:パブコメ)が求められる。パブコメの結果を踏まえて、EWGによりGL案が修正される。
【Step4】
GL案が総会の規制当局の代表者によって最終的に合意され、採択される。
※ICHの役割としてはここまで。
【Step5】
ICHの各地域・国の規制当局において、それぞれの手続きにしたがってガイドラインが実施される。日本では、厚生労働省医薬・生活衛生局から通知される。
この中で、Step2bが完了した後にGL案が公開されるタイミングとGLが完成するStep4のタイミングで注目度が非常に高まります。
私は、Step2での情報が特に重要だと考えていて、どのような内容がGLに入ってくるのかが明確になってくることから、Step5までにどのような準備をしなければいけないのかが分かりますし、それに付随して医薬品開発においてどのような需要が発生しそうかが掴めるタイミングだと思っています。
その他
その他、ガイドラインには「(R●)」や「Annex●」(●には数字が入る)が付いていることがあります。
Rは、Revisionの「R」で改訂を意味しています。そのため例えば、「E6(R3)」という表記であったら”E6ガイドラインの3回目の改訂”ということになります。
また、Annexは付属文書という意味合いなので、Annex単独では存在しません。
ICH E6(R3)とICH E8(R1)
とICH-E8(R1).jpg)
今回は、ICH即時報告会の中でもCRCさんやCRAに影響するE6とE8をご紹介するとお話をしましたが、簡単にではありますが、それぞれがどのようなガイドラインなのかについて触れておきたいと思います。
正直、臨床試験に関わる方にとってこのE6とE8は超重要ですので、しっかりと押さえておきたいところです。
ICHの内容が難しくて拒否反応がある方でも、何とか分かりやすいように書いてみましたので、是非ご覧下さい!
ICH E6とは?

ICH E6は、「医薬品の臨床試験の実施基準」に関するガイドラインでいわゆるGCPのことです(業界外の方は、GCPについては、”治験を実施する上で守らなければいけないルール”のように思ってもらえればOKです)。
治験に携わるのであれば、誰しもが必ず学ばなければいけないGCPの大元がこのICH E6(「R1」と付いてはいますが、これは新コードであり、旧コードはE6でした)ということになります。
そして、日本では1997年3月27日(平成9年)に「医薬品の臨床試験の実施の基準に関する省令の施行について」という通知が厚生省薬務局長通知として発出されました。
これが「新GCP」の誕生ということですね。
その後、ICH E6(R2)が2019年7月5日(令和元年)にSTEP5に到達し、現在まで運用されています。
このICH E6(R2)では、主に品質マネジメントとRisk Based Monitoring(RBM)についての考え方が示され、今までは出口管理であったモニタリングが、プロセス管理に進化し(品質マネジメント部分)、リスクの評価に応じてモニタリングの強弱をコントロールする(RBM部分)よう進化をとげました(実際は、進化できていないこともあるんですけどね…)。
そして、今回の即時報告会で報告があったのがICH E6(R3)についてですので、ここから更なる進化を今後とげることになるということです!
実際に報告された内容は後述します。
リスクに基づくモニタリング(Risk-Based Monitoring:RBM)という考え方は、2011年8月にFDAとEMAにより相次ぎ発表されたガイダンス案が始まりでした。 その後、2013年には日本でも「リスクに基づくモニタリングに関する基本的な考え方について」が発出され、2015年にはICH-E6 Step2ガイドラインに掲載されました。 今までもよりも適切で効率的なモニタリング手法として考案されたRBMですが、昨今のCOVID-19の蔓延もあり、再度注目が高まってきています。 今回はそんなRBMについて、基礎的なところからお話していきたいと思います。(意外に単純なので、就活生や経験が浅い方も理解できると思いますよ!)

ICH E8とは?
ICH E8は、「臨床試験の一般指針」で、1998年4月21日にSTEP5に到達して作成されました。
E8では、医薬品開発を行うための開発戦略や臨床試験に関する原則、関連用語の定義などがまとめられており、それぞれの詳細な内容については、E8以外のガイドラインを参照するよう促すガイダンス的な役割を担っています。
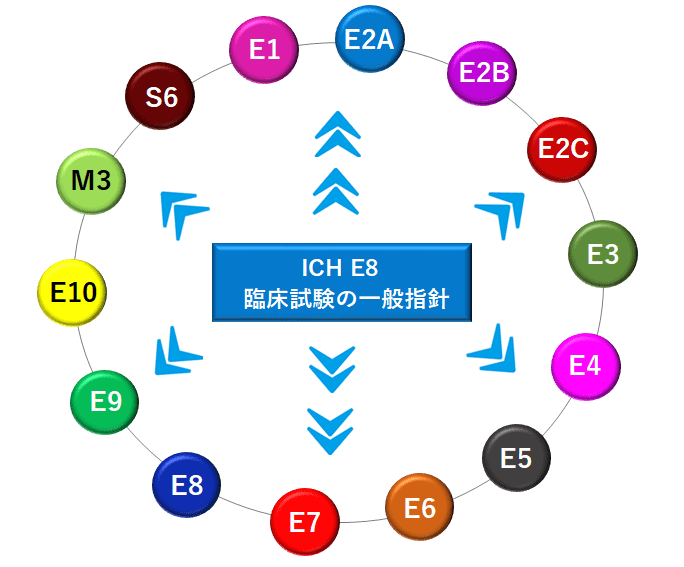
ガイダンス的な役割を担っていることから、臨床試験に関わる際には、まずICH E8から読み始めて、詳細については、参照されている各ガイドラインで確認する手順で見ていくと良いです。
CRO/製薬メーカーの新人研修(SMOはよく分かりませんが)では、CRAスタートの場合、GCPから学び始めることも多いのですが、個人的にはICH E8をまず読んで、それからGCPを読んで順序の方が良いと思っています。
なにせ、E8では、GCPであるE6への道しるべもあるのですから。
以上より、ICHは現在E8を改訂し、その後、E8に紐付くE6を改訂するという順序で改訂が進んでいます。
また、現在はICH E8からのガイドはありませんが、国際共同治験に関わる方は、ICH E17である国際共同治験についてのガイドラインの確認は必須ですので、該当する方は是非ご確認を!
第43回ICH即時報告会

冒頭でも述べた通り、2021年7月7日には、第43回ICH即時報告会が開かれ、ICH E6(R3)やICH E8(R1)などの進捗報告がされました。
上記以外にも、ICH M8のeCTDについて等の報告もされましたが、今回は現場で働く方向けということで、ICH E6とE8に絞って概要をまとめていきます。
また、先ほども上述した通り、ICH E8はガイドライン的な役割を担っているということもあり、こちらでの概要報告もE8から先にまとめています。
ICH E8(R1)
ICH E8は、1998年に作成されたわけですが、それから20年以上の月日が経ち、医薬品開発の環境も随分と変化してきました。
細かく見ると色々な改訂内容があるのですが、その中でも押さえておくべきポイントは、”質を試験の計画時点で作り込むQuality by Designについて”と”患者の意見を医薬品開発にどのように活かしていくのかというPatient Centricityについて”が追加される点です。
Quality by Design
E8(R1)の目次に新たに”DESIGNING QUALITY INTO CLINICAL STUDIES(臨床試験における質の設計)”が加わります。
中身は、”Quality by Design of Clinical Studies(臨床試験におけるクオリティ・バイ・デザイン)”、”Critical to Quality Factors(質に関する重要な要因)”、”Approach to Identifying the Critical to Quality Factors(要因を特定するアプローチ)”で構成されます。
Quality by Design(QbD)についての説明にあたる「Quality by Design of Clinical Studies」では、QbDに必要な項目として以下のことが述べられています。
●the need for clear pre-defined study objectives that address the primary scientific question(s);
●selection of appropriate participants that have the disease, condition, or molecular/genetic profile that is being studied;
●use of approaches to minimise bias, such as randomisation, blinding or masking, and/or control of confounding;
●endpoints that are well-defined, measurable, clinically meaningful, and relevant to patients;第43回 ICH即時報告会より
上記を理解するための大前提として、ICHで語られる”臨床試験の質”とは、”目的への適合性(Fitness for purpose)”として定義されていることを念頭に置かなければいけません(「Fitness for purpose」は今後あらゆるところで耳にすることになると思いますので、しっかりと定義を把握しておいた方が良いです)。
そのうえで、上記を読み解いていきますと…
試験の計画時においては、事前にしっかりと明確な研究目的を設定する必要があり、その目的に合ったプロファイルを持つ適切な参加者(被験者)を選択する必要があるということが意図されていることが分かるかと思います。
また、バイアスを最小限に抑える必要があるため、無作為化や盲検化や交絡因子の制御などが求められます。
その他、エンドポイントについては、適切に定義され、測定可能なエンドポイントと臨床的に意味があり、患者に関連するものとすることが記載されています。
言われてみれば当たり前のことばかりなのですが、現時点では、これらに関しても具体的にしっかりと指針に記されていないため、その目線を明確にharmonizeするためにICH E8(R1)として改訂が進んでいるとうことです。
ちなみに、”測定可能であり、臨床的に意味があり、患者に関連するものをエンドポイントとする”という部分について、私はCRO時代に色々な試験を担当してきましたが、曖昧だなと感じる試験は実際にありました。
例えば、ある検査値Xのベースラインからの変化量をエンドポイントに設定している場合、それが変化することによって臨床的にどのような意味があり、具体的に患者にどのようなベネフィットがあるのかまで考慮する必要があります。
もちろん、考慮だけはどの試験でもされているはずですが、そのうえで、設定したエンドポイントが本当に適切なのかが重要になります。(実際の試験計画の際には、正直なところ、開発コスト的なことや開発期間についても社内的に考えなければいけないので、なかなか理想通りに綺麗にいかないこともあるとは思うのですがね…)
今回のICH即時報告会では、詳細には言及されていませんでしたが、Quality by Designは、ICH E6(R2)で触れられたプロセス管理がしっかりと出来ていることが前提となります。
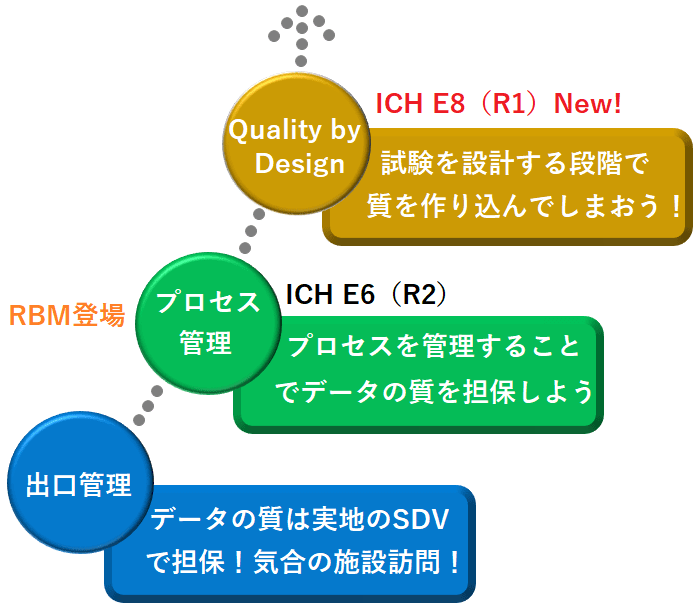
Quality by Design+プロセス管理がしっかりと出来た時、現場でのモニタリングのあり方/考え方などに変化が生じていくのだろうなと思っています(モニタリングでの確認範囲、確認方法、確認の観点の変化など)。
かつては出口管理で、エラーが出たらSDVで見つけ出し修正をすれば良いという考えでしたが、時は流れて”そもそもミスが起きない方法を”という考え方に変化していっているということですね。
また、ここまでの資料では出ていませんが、臨床試験の質はあくまで適切な試験デザインを構築することと(QbD)、それの適切な遂行すること(プロセス管理)に依存すべきであり、後方視点的なモニタリングや監査や査察などに過度に依存すべきではないとされています。
ここまでは、QbDとして、依頼者側の質の作り込みについて触れてきましたが、医療機関側での質の作り込みであるBuilt-in qualityも大変重要なワードとなりますので、QbDと併せて覚えておくと良いかと思います。
Patient Centricity
E8(R1)の”GENERAL PRINCIPLES(主原則)”に「Patient Input into Drug Development(医薬品開発における患者情報)」の項目が加えられます。
説明としては、以下のように説明がありました。
Consulting with patients and/or patient organisations during drug development can help to ensure that patients’ perspectives are captured.
第43回 ICH即時報告会より
あまり詳細に詳しくは語られていませんでしたが、Patinet Centricityについては、最近では医薬品開発の場ではよく聞くようになってきた考え方で、正式にICHのGENERAL PRINCIPLESに乗ることはとても意味のあることだと思っています。
ざっくりと患者さんの意見をどのように医薬品開発に取り入れていくかということについては、こちらの資料を読むと分かりやすく記載されているのでおすすめです。
患者さんの声を医薬品開発に活かす方法は色々とあります。例えば、治験に参加する際の同意説明文書の作成に患者さんの意見を反映していこうという取り組みもそうですし、患者会にコンタクトを取って患者さんの声を聞くことも含まれます。
実際には、サイエンスの視点から作成された治験のプロトコールに患者さんの意見を取り入れることは難しいことが多いのですが、難しいからと言って患者さんの意見を切り捨てるのではなく、しっかりとどのような形であれば意見を取り入れられるかを模索していくことが求められます。
進捗
COVID-19の影響で進捗が遅れているようですが、近々STEP4には到達予定とのことです(この夏にも)。
STEP5についていつ頃になるのかという質問については、「速やかに対応します」とのことで、明確な答えが無く、次回の進捗報告が待たれるところですね。
ICH E6(R3)
ICH E6はいわゆるICH GCPのことです。
ICHの方向性を記した2017年1月のReflection Paperには以下のように記載されています。
1: Revision to ICH E8
•Address broader concerns about the principles of study design and planning for an appropriate level of data quality
•Provides comprehensive cross-referencing to the family of ICH guidance documents2: Renovation of ICH E6 GCP
•Address flexibility concerns with respect to a broader range of study types and data sources
•Retains the current focus on good clinical investigative site practices
最近では、臨床試験のデザインやデータソースの多様化が進んできていることから、ICH E8を刷新して、GCP Renovationとして、ICH E6ガイドラインも改訂することとなりました。
そして、ICH E6(R3)の全体の構成は以下のようになります。
の構成.png)
紫色の四角い枠がICH E6の本体部分で、別添としてAnnex-1とAnnex-2があります。
Annex-1は、ICH E6の概念を反映しており、結びつきが強いため、E6(R3)の改訂においても本体と同じ動きで進捗しており、本記事を執筆している2021年7月12日現在では、11月のSTEP1/2到達を目標に進めている段階のようです。
また、Annex1に記載されていないnon-traditionalな追加事項を記載したAnnex2はAnnex1がSTEP1/2に到達後検討が開始されるとのことなので、具体的な内容が見えてくるのは2021年11月以降になりそうです。
ちなみに、Annex1とAnnex2の円が交わる部分には、Pragmatic TrialsやDecentralized Clinical Trial(バーチャル治験とも言う)やReal World Dataなどが含まれており、どれも今後増えてくることが想定されている注目の内容が含まれます。
これらはAnnex1で語られますが、医薬品開発業界ではこのトレンドを敏感に察知して先行して準備に取り掛かっている企業もあり、開発者の立場として、具体的にどのような未来になるのかがとても楽しみです(実現可能性や影響度を加味して関連企業に投資を始めたりもしています!)。
このICH E6(R3)は、STEP2に到達していないためGL案が発表されておらず、具体的な内容は現時点ではありませんが、方向性については、即時報告会でも報告されていました。
Data Management
今までのE6では語られていなかったウェアラブルやAI等の新しい技術/データについての言及やITセキュリティ、ユーザー管理について触れられるようです。
最近では、色々な技術が発達してきたので、確かにリノベーションが必要な項目でしょう。
Responsibilities
こちらでは、DCTにおける治験責任医師と依頼者の責任の所在についても触れられるようです。
確かに今までの治験とは違い、治験行為をするところが治験実施医療機関のみではなく分散された場合に、治験責任医師の責任の範囲がどこまで及ぶのかは個人的にかなり気になっていましたが、明確なラインが引かれるとのことで、詳細がより気になります。
DCTといっても色々な形態をとることも想定できますので、どのようにまとめられるのでしょうね。
その他、契約書や合意書への記載事項やCROなどのベンダーの事前評価、実績評価についても触れられるようです。
Feasibilityにも関連する内容の更新があるのかと思っています。
Monitoring
かつてよりEDCが急速に普及し、更に様々なタイプのモニタリング(例えば、リモートモニタリングやセントラルモニタリングなど)が実施されるようになってきたため、それらについて言及する内容になるようです。
ソースデータ及びメタデータのレビューとソースデータの検証、サイトシステムのレビューやその他の適格性確認のための要件の明確化などが挙げられており、モニタリング時に確認しなければいけない個所やモニタリング自体に対する考え方に追加事項があるような印象を持ちました。
進捗
先ほども少し触れましたが、Principles+Annex1はSTEP1/2到達が11月以降、Annex2の検討開始は更にそれ以降になります。
現時点では、GL案が見れる状況にはありませんが、年末頃に見れたら早い方なのかなと思っています(やはりコロナの影響で遅れそうなので…)。
まとめ
今回は、ICHの基礎から、ICH E6、E8についての基礎知識と、先日参加した第43回ICH即時報告会での概要にについて簡単にまとめていきました。
今回は長くなり過ぎたので、本文では触れませんでしたが、日本発としてRWD関連のガイドライン作成の提案もし、採択されています。
日本発で質の高い提案をすることは、世界での存在感を示すためにも非常に重要なことなので、今回の採択もとても大きな意味を持つもので、今後の進捗が非常に楽しみです。
また現在は、日本における国際共同治験の治験届で件数もかなり増加しており、今後は日本で治験をやるとしてもICHを知らずして進むことはできない状況になってきます。
医薬品開発全体を知るのであれば、ガイドラインを一通り読むのが良いかと思いますが(特殊なものもありますが…)、新人の方やこれから開発業界で働く方は、まずは自分に関係がある部分のみ見てみるのでも良いかと思います。
GCPがリノベーションされ、古い考え方から切り替わるタイミングは近いので、是非乗り遅れないように最新動向をチェックしていきましょう!(私も頑張ってチェックします!)






“ICHの簡単な概要とE6(R3)・E8(R1)の概要まとめ/第43回ICH即時報告会” への1件のフィードバック