

医薬品開発業界には様々な専門用語が登場します。
更に厄介なことに、治験に登場する用語は英語の略語も多く、慣れないうちは何のことを示しているのかが分かりにくいという特徴があります。
そこで、今回は、治験で登場する用語(特に現場で登場する用語)について、簡単な解説付きで紹介をしていきたいと思います。
なお、各用語について、会社毎によって微妙にニュアンスが違うこともあるかと思いますので、本記事では大まかなイメージを持っていただくことを想定し、記事を書かせていただいています。
役割や組織の名称

このカテゴリーでは、治験での登場人物や組織の名前にフォーカスして、比較的よく登場する用語をまとめてみました。
用語単独ではイメージしにくいかと思い、それぞれの関係についても図式化していますので、併せてご確認下さい。
CRO
CROとは、Contract Research Organizationの略で、日本語では医薬品開発業務受託機関と呼ばれている(ただし、日本語名称は現場の会話の中ではほとんど登場しない)。
CROは、主に製薬メーカーや医療機器メーカーを顧客とし、臨床開発業務(モニタリングなど)を受託し、サービスとして提供する会社である。
受託している業務の幅は会社によりそれぞれだが、大手になるとほとんどの臨床開発業務を受託することができる体制が整っている(トータルソリューションという形でアピールをしているCROも存在する)。
CROに所属するCRAは、医療機関と依頼者(製薬メーカー、医療機器メーカー)の板挟みに合うことも多々あり、立場的に業務を進めにくい場面もある一方、多くの依頼者の開発に携わることが出来る為、幅広い経験を積むことも出来る。
「この依頼者カオス」、「この医療機関カオス」という会話は日常茶飯事である。
近年医薬品開発業界においてその存在感が日に日に増しているCROですが、今後の将来性についてどうなのでしょうか? この記事では、CROと製薬メーカーどちらにも所属した経験がある私の考えを紹介していきたいと思います。 医薬品開発業界は日々進化を続けており、その進化にしっかりと着いていくことが出来るかが大きなポイントです。 最近の医薬品開発の現場のトレンドにも触れながらCROの将来性について一緒に考えるきっかけになれば嬉しいです。

CRA
CRAとは、Clinical Research Associateの略で、治験依頼者側の立場で仕事をする職種である。
主な業務内容は、治験実施医療機関において治験が規定通りに適切に実施されているかをモニタリングする役割を担っている。その為、CRAはモニター(臨床開発モニター)と呼ばれることも多々ある。
CRAは、モニタリングの他にも、治験実施医療機関においてスタッフ向けに治験の説明や治験に関連する資料の作成等の業務も行うため、意外に内勤業務も多い。
余談だが、飛行機を多く使用するCRAは、マイルも多く貯まるため、上位ステータス保持者が多い。
上位ステータスになると専用ゲートからの入場が出来るため、空港でドヤ顔が出来るのもCRAの旨味である。
また、上位ステータスであることを良いことに、ラウンジでくつろいでいるCRAも多いことからラウンジで「C☆R☆A」と口ずさめば何人かは振り返り共に口ずさむという状況も考え得る。
その中にのりすもいる可能性があるため、是非試してほしい(嘘)。
就職活動の段階では、色々な職業の話を聞いてみてもその将来性まではなかなかイメージできないもの。それは、臨床開発職であるCRAについても同様かと思います。 そのような状況でも、色々な職種の中からどの職種になるのかを考えるときにその職種の将来性を考えることはとても重要なことです。 本記事では、CRAの業務を経験した立場からCRAの将来性についてお話をしていきたいと思います。

CRAは専門的な知識を生かして治験がしっかりとおこなわれているかを確認するお仕事になります。 その他、治験が円滑に行われるために医療機関の関係者に治験の説明をしたり、治験資材を搬入したりと幅広い業務を担っており、CRAに …

QC
QCとは、Quality Controlの略で、治験依頼者側の立場で仕事をする職種である。
主な業務内容は、治験で発生する様々な書類の品質(誤記が無いか、正しく資料が作成されているか等)を確認することである。
CRAのモニタリング報告書をレビューすることもあり、CRAが不甲斐ないと怒りの鉄槌が飛んでくることもある。
あまりにも仕事が雑なCRAは、QCから目を付けられてしまうため、CRAとしてはクオリティの高い資料作成を心掛ける必要がある。
QCは、内勤業務の為、CRAとして勤務していた方が産後の復帰先として選択することも多く、女性の割合が多い。
QCから好かれているCRAは、“できるCRA(通称イケメンCRA)”である割合が高い。
なお、QCという単語は職種以外にも動詞的な使われ方もすることがある(例:この資料はQCされたものだが、クオリティがズタボロのようだ)。
DM
DMとは、Data Managementの略で、治験依頼者側の立場で仕事をする職種である。
主な業務内容は、EDC(詳細は後述)の管理や保守である。
治験のデータがEDCに入力されるが、データの入力内容に疑義事項がある場合には、クエリ(問い合わせ事項)を発出したり、CRAに問い合わせをすることがある。
DMにしっかりと治験の取り決め等を共有しておかないと、施設の方がDMが発出したクエリに対して激おこすることもあるため、CRAとしてもしっかりとDM担当者とコミュニケーションを取っておく必要がある(直接CRAが関われない場合は、PL(Project Leader)等に依頼をする必要がある)。
SMO
SMOとは、Site Management Organizationの略で、日本語では治験施設支援機関と呼ばれている(ただし、日本語名称は現場の会話の中ではほとんど登場しない)。
SMOは、治験実施医療機関と契約をして、治験関連の支援業務をサービスとして提供する会社である。
SMOに所属するCRCもまた、医療機関と依頼者(CROを含む)との板挟みに合うこともあり、柔軟な立ち回りが求められる。
CRAは、全国の医療機関を担当する可能性があるが、SMOのCRCは基本的には所属する支店が管轄しているエリア内の医療機関を担当するため、全国的に移動することはほとんど無い。
SMOは、医療機関での治験支援サービスの提供の他に、治験実施医療機関となり得る病院・クリニックを開拓していくことも行っている。
メーカサイドも医療機関サイドも色々な職種があるが、なんとなく以下のようなイメージである。
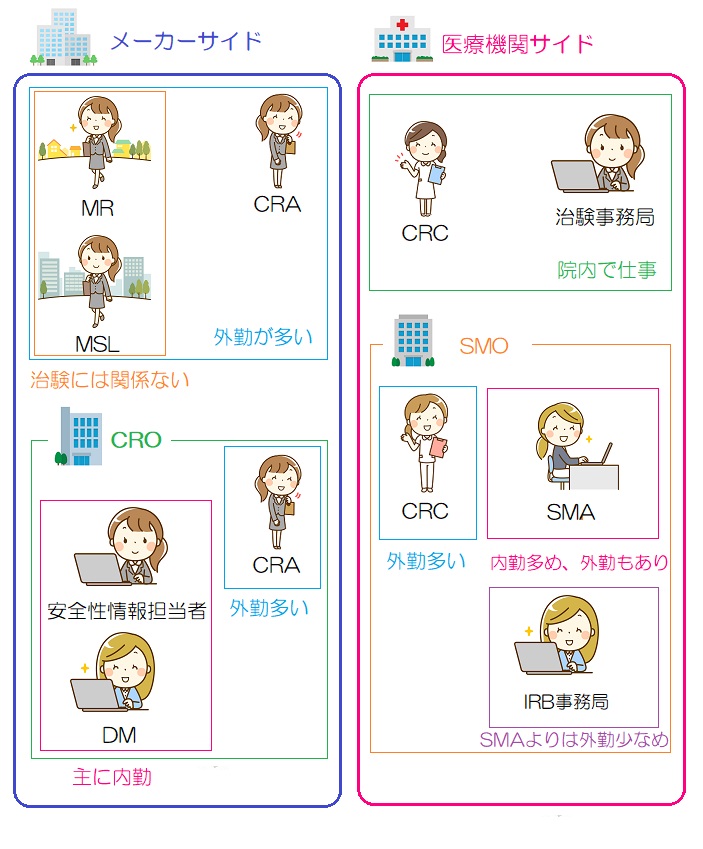
CRC
CRCとは、Clinical Research Coordinatorの略で、医療機関側の立場で仕事をする職種である。
主な業務内容は、治験実施医療機関において治験が円滑に実施出来るよう医師を始めとする各医療機関スタッフの支援をし、治験をコーディネート(調整)する役割を担っている。その為、治験コーディネーターと呼ばれることも多々ある。
CRCは、被験者へ治験の補助説明をしたり、治験に関しての補助資料を作成したり、治験に参加できそうな候補者がいないかのカルテスクリーニングをしたりと医療機関での治験関連業務を幅広くこなしている。
CRCには、SMO(詳細は後述)に所属するCRCと医療機関に所属する院内CRCがいる。
CRCは、CRAの対応次第で、最大の助っ人とも最大の壁ともなり得る。CRAとしては、誠実・適切・迅速な対応を心掛ける必要がある。
なお、経験上、”気が利くCRA”は人気度合いが高い気がする為、その点も留意する必要がある。
SMA
SMAとは、Site Management Associateの略で、日本語では治験事務局担当者と呼ばれている医療機関側の立場で仕事をする職種である(ただし、日本語名称は現場の会話の中ではほとんど登場しない)。
主な業務内容は、治験関連資料の保管や整理などであるが、SMOのSMAの場合は、新たな支援施設の開拓業務に当たることもあるようだ。
ちなみに、治験事務局は、「治験事務局」と「IRB事務局」と分けて呼ばれることもある。
その違いは、医療機関で保管すべき資料を管理しているか、IRBで補完すべき資料を管理しているかの違いである。
場合によっては、治験事務局がIRB事務局を兼ねていることもあるため、治験開始時にはその辺りの運用も確認しておく必要があるだろう。
長(ちょう)
長とは、実施医療機関の長の略で、長すぎる為、短縮して呼ばれることが多い。
役割として「実施医療機関の長」というものがあるが、実際には、事務局担当者に業務を依頼しているため、長自らが対応することはほぼ無い。
「この資料は長から依頼者ですよね?」、「これは長宛の資料です」というように、書類の流れを説明する場面でよく使われるワードである。
なお、資料の流れを説明する際には、後ほど登場する「治験責任医師」のことを責と短縮して呼ぶことも多い。
「長、責、依頼者、IRB」の4者について覚えておけば大体の書類の流れはフォローできるはず。
依頼者は「依」と短縮するケースはほぼ聞かないので、勢い余って依と略すと何とも言えない空気になり恥ずかしい思いをするかもしれないが、あえてボケをかますというのも一考の余地がある(なぜ「依」と略さないかは解明されていない)。
PI
PIとは、Principal Investigatorの略で、治験責任医師のことを指す(現場では「PI」や「責任医師」と呼ばれている)。
PIは、治験を担当するスタッフを指名したり、治験で生じた有害事象(詳細は後述)の評価をしたり、被験者へ治験の説明を行ったりと多くの役割を担っている。
治験関連の業務については、CRCが広くサポートをしており、PIは医学的判断を伴う部分(有害事象における因果関係の判定や合併症の判断など)を主に対応している。
また、治験のデータをまとめる為に使用するワークシート(詳細は後述)の記載にも対応をするが、忙しいPIにとっては負担となる業務の1つである。
なお、PIには診療部長など、偉いポジションの医師が就くことが比較的多い(クリニックの場合は、院長がPIとなる場合が多い)。
SI
SIとは、Sub Investigatorの略で、治験分担医師のことを指す(現場では、「SI(SubI)」や「分担医師」と呼ばれている)。
SIは、治験に関わる頻度が高い先生と低い先生(ほぼ関与しない先生)がいるため、key Dr.となるSIを把握しておくことが重要である。
なお、PIが診療部長等偉いポジションの先生が就いており、SIがその下の先生である場合、無理矢理仕事を振られていることもあり、PIのモチベーションが高くてもSIのモチベーションが低い場合もある。医師の世界も大変なのである。
SIが担当医師となっている患者が被験者となる場合には、有害事象の判定やワークシートへの記載をSIが担当することになる。
また、関りがほとんどないSIの場合は、気が付いたら異動や退職していることもあり、一定期間ごとに気を配る必要があるだろう。
IRB
IRBとは、Institutional Review Boardの略で、治験審査委員会のことを指す。
IRBは、専門委員と非専門委員により構成されており、治験実施医療機関において、治験を実施することの適否を審議したり、新たな安全性情報が出た際に、治験継続の可否を審議したりと治験関連の審議をおこなっている組織である。
大学病院などの規模が大きめな医療機関の場合は、自前でIRBを持っていることが多く、自前のIRBを使う際には、「院内IRB」という単語がよく出てくる。
また、自前のIRBを持っていない医療機関は、他の医療機関やSMO等が支援している外部のIRBを使用することがあり、「外部IRB」という単語がよく出てくる。
なお、複数の施設で治験を実施する場合には、1つのIRBでまとめて審議にかけることもあり、そのような形態のIRBを「セントラルIRB」と呼ぶ。
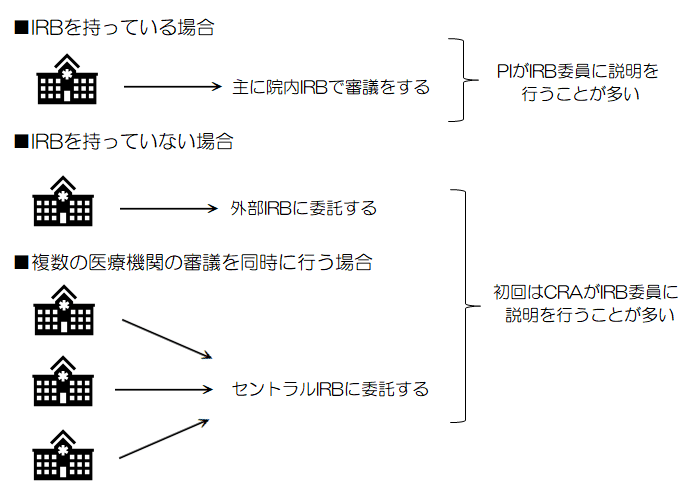
治験を実施することについての審査(通称:初回審査)では、CRAがIRB委員に発表(治験の概要等)をしなければいけないこともあり、緊張に弱いCRAにとっては鬼門の1つとも言えよう。
IRBの委員には色々な方がおり、想定していた質問外からのアッパーを食らうこともあるため、高度な適応力が求められる。
その為、CROのCRAがIRBで発表することになったときは、心の中で「依頼者、お願いだから同行しやがって下さい」と思っている事だろう。
なお、会話の内容は議事録に残るため、適当なことを言うのはご法度である。
PMDA

PMDAとは、Pharmaceuticals and Medical Devices Agencyの略で、独立行政法人医薬品医療機器総合機構のことを指す。また、「当局」と呼ばれることも多々ある。その他、愛称として、パンダと呼ばれることも。
PMDAは、「審査」、「安全」、「救済」という3つの役割を担っているが、治験においてはそのうちの「審査」で主に関わることになる。
具体的には、治験のデザインについての相談を受けたり、治験の結果を審査している。
CROのCRAが関わることはほぼ無く、PMDAとバトルをし合う連絡を取り合うのは主に依頼者になる。
また、医薬品の審査をする過程で、依頼者と医療機関に査察が入るため、CRCは当局の査察官の対応をする場面もあり、緊張に弱いCRCにとっては鬼門の1つとも言えよう。
実務でよく使う用語・略語

ここでは、実際に実務をしていくうえでよく見かけたり、耳にする用語や略語をまとめてみました。
本当は、もっと色々あるのですが、比較的出現率が高めのものor重要なものを紹介しています。
GCP
GCPとは、Good Clinical Practiceの略で、「医薬品の臨床試験の実施の基準に関する省令」のことである。
GCPには、治験を実施する上で遵守すべき事項が規定されており、CRCやCRAなど治験に関わる人(本当はPIやSIもだが実際は…)についてはこのGCPを熟知している必要がある。
なお、CRCやCRAはGCPをいつでも参照できるように、みんな大好きポケット資料集を持参している。
ポケット資料集という単語が出てきた場合には、120%の確率でGCPの何かについて語っている時である。
なお、ピンク色のポケット資料集を華麗に使いこなしているCRAのイメージはパレクセルのHPにイケている画像があるので、就活生などはこれを見てイメトレをして欲しい。付箋が貼ってあるところあたりがリアルである。

採用情報|パレクセルより抜粋
SUM
SUMとは、Start Up Meetingの略である。
新たに治験を施設で始める際には、治験の概要や各スタッフに対応していただきたい内容をCRAが発表する。
関係者を1ヵ所にまとめて大人数で開催する場合もあれば、薬剤科、検査科など、部門ごとに区切って複数回行う場合など、色々なパターンがあるが、これらのことをまとめてSUMと呼ぶ。
SUMが開催されるタイミングは、様々だが、SUMが開催された以降に被験者の組み入れが始まる。
IRBでは、斜め上から攻撃が来るのに対し、SUMでは医療機関のスタッフからの質問であるため、真正面からの正拳突きがやってくる。
アプローチは違えど、緊張に弱いCRAにとっての鬼門の1つであると言えよう。
また、SUMと似たような用語でSIV(Site Initiation Visit)というものがある。
SUMと似たように使用されることもあるが、SIVは正確には治験の準備が全て整い、最終的な打ち合わせを行うために医療機関へ訪問することを指している。
つまり、SUMをした翌日に治験が開始出来なくても問題は無いが、SIVの翌日には治験が開始できる状態でなければいけないということである。
なお、SUMは広く使われている一方、SIVという言葉は、外資系メーカー・CROで比較的使用されており、一部の内資系メーカー・CROでは馴染みが無いCRAもいる。
CRC勉強会
CRC勉強会とは、ある施設で治験が開始する前にその施設のCRCを対象にCRAが行う勉強会である。
CRC勉強会では、治験の概要はもちろんのこと、実際にどのような運用で進めていくのか詳細をしっかりと詰めていくことが目的であるため、CRAの理解があやふやなままCRC勉強会を実施してしまうと怒りの鉄拳が飛んでくる可能性がある。
CRC勉強会を実施するタイミングは、IRBで治験が実施することが確定した後~スタートアップミーティングがほとんどである。
なお、CRC勉強会を実施するタイミングは、治験の立ち上げの時期であるため、CRAは忙殺されており、色々な運用が頭に入り切っていない状態でCRC勉強会を迎えてしまっているCRAもしばしば見られる。
私の経験上の話だが、CRC勉強会の冒頭で、どのようなベンダー(EDCやWeb登録センターや検査会社等)を使うかをまず始めに説明した後により詳細な内容の説明に移る方がスムーズに話が進むことが多い。
ベンダーを先に話すことで、CRCがある程度運用をイメージしてくれるので、CRAはその治験のイレギュラー部分に注力をして話すことでしっかりと相手に着目ポイントを伝えることが出来るだろう。
CRAをある程度経験していけば分かるが、運用を全て頭に入れようとするのは至難の業であり、実際のところは押さえるべきポイントをしっかりと押さえて運用を頭に入れることで効率良く運用を理解することが出来る。
その辺りの感覚は経験によって鍛え上げられるものである。
AE
AEとは、Adverse Eventの略で、有害事象のことを指す。
有害事象とは、治験使用薬又は製造販売後臨床試験薬を投与された被験者に生じた全ての好ましくない又は意図しない疾病又はその徴候(臨床検査値の異常を含む。)のことである。
基本的には、AEは、治験薬服用期間中に生じた事象のことを指すが、プロトコールによっては、治験薬服用後の後観察期間までAEの情報を収集する期間として規定されている(最近はかなり多くなってきたように思う)ので、新しいプロジェクトにアサインされてプロトコールを確認する際には、しっかりと押さえておきたいポイントだ。
SAE
SAEとは、Serious Adverse Eventの略で、重篤な有害事象のことを指す。
AEのうち、以下に該当した場合はSAEである。
●死亡
●死亡のおそれ
●入院又は入院期間の延長
●障害
●障害のおそれ
●上記に準じて重篤
●先天異常
SAEは発生した際には、治験責任医師がその情報を知ってから24時間以内に治験依頼者に報告する必要があり、土日あるいは、帰宅間際に発生した場合には発狂ものである。
ほとんどの場合は、「入院又は入院期間の延長」でSAEとなる。
非常に緊急を要する対応のため、CRC、CRAともに負担が大きい対応である。
SAEを頻繁に引き当ててしまうCRC/CRAについても”もっているCRC/CRA”と呼ばれることがあり、よく引き当てる人は引き当てる一方で、全く引き当てない人もいる。
なお、”激しくもっているCRC/CRA”は、年末年始やGWにも引き当ててくる。
SAEの発生頻度は、担当する疾患領域に大きく依存するため、抗がん剤関連の開発に携わる場合にはそれなりの覚悟が必要である(対応しすぎて慣れるので恐れすぎる必要は無し)。
ADR
ADRとは、Adverse Drug Reactionの略で、副作用のことを指す。
つまり、有害事象のうち、因果関係を否定できない反応のことである。現場では、ADRという単語よりは、普通に「副作用」という言葉を使うことが多い。
有害事象(AE)と副作用(ADR)の関係は以下の通りである。
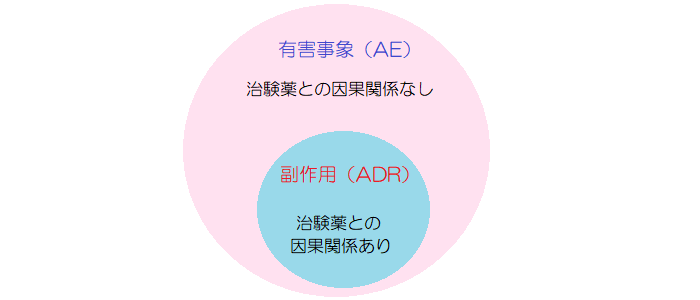
書式〇
治験で扱う書類に統一書式というものがある。
この統一書式というのは、治験を実施する上で発生する様々な手続きを行う際に発生する書類のことであり、どの治験でもなるべく統一した書式を使用することで、効率的な手続き対応にしていきましょうという目的で制定されたものである。
統一書式には、正式名称があるのだが、それぞれの書式にはナンバリングもあり、正式名称を言うのが面倒なときには「書式○」など、省略して呼ばれることがある。
それぞれの書式のナンバリングが頭に入っていないと全く会話に着いていけないため、経験が浅いCRC、CRAにとっては混乱をきたす恐れがある。
治験実施中にも比較的出現頻度が高い統一書式を一部まとめてみた。とりあえず、この辺りは頭に入れておきたい。
●治験審査結果通知書(書式5)
●治験に関する変更申請書(書式10)
●重篤な有害事象に関する報告書(書式12)
●安全性情報等に関する報告書(書式16)
ちなみに、書式○以外にも、統一書式は他の略し方で呼ばれることも多々ある。
上記の資料についてその略し方でよく耳にするものを挙げておく。
●治験審査結果通知書⇒結果通知書
●治験に関する変更申請書⇒変更申請書
●重篤な有害事象に関する報告書⇒SAE報(速報、詳細報)
●安全性情報等に関する報告書⇒安情
個人的には、書式○と言うと、伝わらないときがあるため、略すとしても「統一書式の略し方」で示しているような、意味合いが分かる略し方の方が良いと思う。
私が新人の頃にアサインされたプロジェクトの先輩は新人の私に「12受け取って、4からの5だろ?それからの16よ」と話されていたが、海外ドラマ・LOSTのファンなのかと感じてしまったことは良い思い出だ。
なお、今でも大規模な病院では、統一書式以外にも院内書式というその医療機関でのみ使用する書式が存在していることもあり、医療機関が毎に柔軟に対応が求められることもある。
WS
WSとはWork Sheetの略である。
WSは、治験の為に収集した情報を記録する用紙であり、治験開始時にはCRAがCRCに電子媒体を提供し、施設がやりやすい形にカスタマイズをして使用することが多い。
カスタマイズは、CRCがやる場合もあれば、CRCからリスクエストを受けて、CRAがおこなう場合もあり、その辺りの立ち回りは、その場の空気を読んで対応することになる。
なお、オラオラ系のCRAの場合は、「わしは絶対やらんぞ」というオーラを放ち牽制を始める人もいるが、後々のコミュニケーションを考えるとお利口さんにしておいた方が良さそうだ。
ちなみに、WSをわざわざ日本語に直して呼んでいるのは聞いたことが無いが、無理矢理直すとしたら「労働用紙」となり、なかなかの味を醸し出している。
WSは、文章で記載するときに使うが、わざわざ「だぶりゅーえす」とは呼ばず、会話では「わーくしーと」と呼ぶので始めて口に出すときには思い出して欲しい。
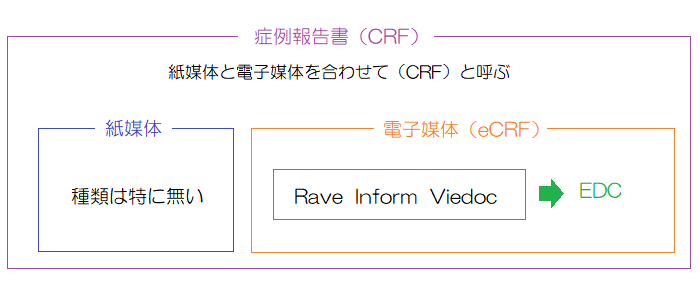
CRF
CRFとは、Case Report Formの略で、症例報告書のことを指す。
症例報告書は、施設(治験実施医療機関)で作成される資料で、治験で収集が規定されているデータを記録する資料である。
最近の症例報告書は、紙媒体から電子媒体に移行してきているため、電子媒体のCRFのことを紙媒体と区別してeCRF(electronic CRF)と表記することがあるが、会話にはほとんど出てこない。
eCRFは「EDC」という用語と非常に関連が強い(詳細は後述)。
ただ、業界では、電子媒体の症例報告書のことを含めて「CRF」と呼んでいることが多いので、症例報告書が紙媒体なのか電子媒体なのかは、前後の文脈で判断する必要がある。
治験に組み入れられる症例数が少ないような治験の場合(第I相の治験など)は、紙媒体のCRFを使用することもあるが、それ以外はほとんど電子媒体のCRFである。
EDC
EDCとは、Electronic Data Captureの略で、治験で収集が規定されているデータを電子的に入力するシステムのことを指す。
なお、EDCというのは、システムの総称のことであり、EDCの中にもRaveやInform等色々な種類がある。
例えて言うと、EDCはスマホ、RaveやInformはiPhoneやAndroidのようなものと考えると少しは分かりやすいかもしれない。
PRT
PRTとは、Protocolを略して表記したものであり、治験実施計画書のことを指す。
治験実施計画書には、選択基準、除外基準、中止基準、併用禁止薬や解析方法に至るまで、多くの情報が記載されているため、CRA、CRCは熟読する必要がある。
PRTの最初の方のページには「緒言」という形で開発コンセプト等が記載されているため、初めてプロジェクトにアサインをされた際には、まずこの「緒言」を読むことをおすすめする。
なお、少し表現が難しいと感じる場合は、経験上、同意説明文書を読んでからPRTを読み始めると理解しやすい。
IB
IBとは、Investigator Brochureの略で、治験薬概要書のことを指す。
IBには、治験薬(今後は治験使用薬)に関するあらゆる情報(非臨床試験の結果、臨床試験の結果、安全性情報等)が事細かに載っている。
実験結果の表やグラフについては、慣れていないと読み解くのも困難である。
なお、このIBは分厚い資料で、中身を全て理解することが出来たら「治験薬オタク」の称号を得ることが出来るだろう。
つまり、IBの中身を全て理解している人はほぼ皆無に等しい。
ただ、CRA、CRCとしては、半減期、ADME(Absorption:吸収、Distribution:分布、Metabolism:代謝、Excretion:排泄)、臨床試験の結果、安全性情報については、なんとなくカバーしておくとカッコイイ。
また、有害事象の「未知」、「既知」の判断はIBに掲載されている事象かどうかで決定されるため、そこだけはなんとしても押さえたいところ。
「治験薬概要書」とそのまま呼ぶ人もいるが、「あいびー」と呼ぶ人もそれなりにいるため、知っていないと何のことだか分からないことになる。
ICF
ICFとは、Informed Consent Formの略で、同意説明文書のことである。
同意説明文書は、一般の方向けに平易なことばで治験のことが説明されているため、新しいプロジェクトにアサインされた際には、まずICFを読むことをおすすめする。
なお、単純に同意を取ることは「ICを取る」のように使われるため、ICFとはあくまで資料のことを指す。
「ICFを取る」というと、同意を取っているのではなく、資料を(手に)取っていることになるため、爽やかに使いこなすためにこの違いを覚えておくと良い。
また、ICFは同意書部分が複写になっているのが普通であるが、ICFを発注する際にミスってしまうと、複写になっておらず無残な結果になるため、慣れないうちは注意が必要だ(大体の場合は、印刷業者が慣れているため、教えてくれるが…)。
ちなみに、印刷の発注をするのはCRA(あるいは内勤サポート)である。
Delegation Log
Delegation Logとは、治験責任医師が誰にどのような治験業務をどの期間割り振るかの記録をするために使用するログのことである。
Delegation Logには、各スタッフの署名(signature)を記載する欄もあることから、Signature Logとしての機能も兼ね備えている。
このDelegation Logは、それぞれのスタッフが治験業務を実施する前に完成していなければならない為、スタートアップミーティング(SUM)時までに内容を固め、SUMで集合したところを見計らい、各スタッフに署名を貰いに行くとスムーズに対応が出来る。
なお、Delegation Logの形式は、依頼者毎によって異なるが、スタンダードな形として、TransCelerateの資料を参照すると良いだろう。
割り振り治験業務(Study Tasks)には、以下のようなものがある。大体の場合は、カスタマイズ可能なはずなので、CRAは治験開始前にCRCとしっかりと内容を詰めていく必要がある。


Delegation Log|TransCelerateより抜粋
このDelegation Logは、ICH-GCPに記載されているものであり、日本のJ-GCPでは作成は義務ではない。
そのため、内資系のCROの場合や海外展開をせず、日本でのみの開発を考えている内資系のメーカーの場合、このDelegation Logを使用しないで治験をやることもある。
外資系CRO、外資系メーカーの場合は、ほぼ確実にこの資料が発生する。
また、Delegation Logは、治験に関わるスタッフが追加・削除となる場合には都度更新していく必要があり、しっかりと管理をしていないと更新漏れとなるため、スタッフの変更情報とリンクするようにExcel等で管理することも重要である。
Training Log
Training Logはその名の通り、トレーニングを実施した際のログのことであり、医療機関が作成するものである。
スタートアップミーティングや、その他、治験を運営する上でトレーニングが必要と思われる事項(例えば、CRCに対しては、EDCの使用方法など)が発生した場合には、都度、このTraining Logに記録を残していく。
治験分担医師やCRCなど、各スタッフが追加された際にはまずトレーニングが必要となってくるため、スタッフの追加とはセットで考えておくと良いだろう。
なお、冒頭で「医療機関が作成するもの」と記載しているが、依頼者側でテンプレートがある場合も多く、実際には、CRA側から各医療機関にTraining Logのテンプレートを渡し、誰に対してどのようなトレーニングが必要なのかを伝達する必要がある。
また、Training Logは治験期間を通じて大量に発生する可能性もあり、よく迷子になる資料のうちの1つなので、注意したい。
Site Visit Log
Site Visit Logとは、依頼者サイドの人(CRA等)が医療機関に訪問した際に残す、医療機関への訪問記録である。
各治験参加施設に置いておき、訪問の度にCRAが日付・署名を記載し、CRCが確認欄に日付・署名を記載する。
なお、時々CRAがSite Visit Logを持ち歩いていることがあるが、あの行動は通常はNGである為、新人CRAはそれが普通だと思い込まないよう注意すると良い。
噂によると、このSite Visit Logはかつて海外でカラ出張をするCRAが問題となったことから誕生した資料のようだが、真相は不明である。
FD
FDSとは、Financial Disclosureの略である。
FDSは、治験責任医師と治験分担医師を対象に作成される資料であり、治験を依頼する製薬メーカー等の株式保有情報など、経済的な情報を調査するための資料である。
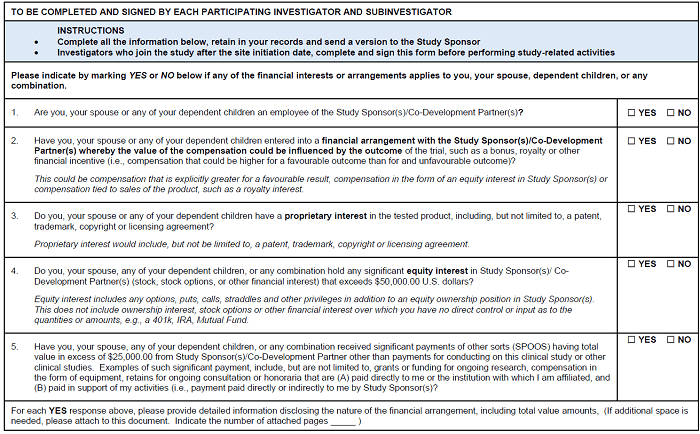
Financial Disclosure|TransCelerateより抜粋
入手時期は、一般的には症例エントリー前であるが(ここがDead Line)、会社毎にいつまでに入手が必要なのかの規定が異なる場合があるため、要確認。
なお、FDについても、日本のGCPには規定されていないため、Delegation Logと同様、日本のみの治験においては発生しない可能性もある。
Form FDA1572
Form FDA1572とは、治験責任医師を対象に入手する資料で、履歴書、治験分担医師・協力者リスト、合意書を1つの資料にまとめたようなものである。
治験責任医師は、このForm FDA1572に署名をすることで、治験依頼者に対して正確な治験の情報を提供すること、FDAの規則に従い治験を遂行することを宣誓することになる。
なお、Delegation Log、FDと同様に、日本のGCPには規定されていないため、日本のみの治験においては発生しない可能性もある。
NTF
NTFとは、Note To Fileの略である。
NTFは、治験実施中に生じたイレギュラー事項について記録として残しておく際に発生する資料である。つまり、イレギュラー事項等が無く治験を終えた場合には、NTFは発生しないこともある。
通常、医療機関側で作成されることが多いが、CROや依頼者側で発生する場合も稀にある。
NTFは決まった形式が無く(プロジェクトによっては指定フォーマットがある場合もある)、使い勝手が良い反面、やたらと何でも記録に残したがる依頼者がいるなど、施設とのトラブルの原因になることもしばしばある資料である。
TMF
TMFとは、Trial Master Fileの略で、治験関連文書のことを指す。
治験では、様々な資料が発生し、発生した資料はTMFに保管されることになるが、「TMF」という用語自体は、外資色が強い呼び方で、実務上では、「責任医師ファイル(治験責任医師が保管すべき資料をまとめたファイル)」や「事務局ファイル」、「IRBファイル」、「依頼者保管用ファイル」等と呼ばれていることも多い。
また、最近では、治験関連文書をクラウド上でやり取りを行うeTMF(electronic Trial Master File)が注目されてきており、このeTMFはRisk Based Monitoringとも親和性が高いため、今後、治験の業界に徐々に浸透していくと思われる。
外資系ではVeevaというeTMFシステムを使用していることが多いが、AgathaというeTMFシステムもCRCあり方会議で共催セミナーを開催する等の取り組みでその存在感を増してきている。
MVR
MVRとは、Monitoring Visit Reportの略であり、モニタリング報告書のことを指す。
モニタリング報告書は、CRAがモニタリングをする度に作成する資料であり、「モニ報」や「モニレポ」と略されて呼ばれることもある(モニ報派とモニレポ派が混在しており、両勢力は拮抗しているように思われる)。
職場で「もにもにもにも」という癒し風な言葉が聞こえてきても油断してはいけない。モニ報は恐ろしいもので、時限爆弾(大体5営業日以内という決まりが多い)が付いており、CRAはこの期限との戦いになるからだ。
なお、MVRと似たような用途で使用するCR(Contact Report)というものがある。
MVRは、基本的には医療機関に訪問した際に作成されるのに対し、CRは、郵送や電話での聴取等、ちょっとした対応の際に作成することが多い。
MVRは、内資でも外資でも存在するが、CRについては、内資では存在しないこともある(全てMVRに記録を残す)。
Confirmation Letter
Confirmation Letterは、医療機関に訪問する前に治験責任医師宛にCRAが通知をするレターのことである。
レターと言っても、手紙を送る訳ではなく、モニタリング報告書の作成ツール(CTMS等)から、電子メールの形で送られることが多い。
医療機関に訪問する度に発生する資料であり、Confirmation Letterを送り忘れて訪問をしてしまうCRAも散見されるため、注意をしておきたい。
また、後述するFollow Up Letterも治験責任医師宛に送るメールだが、通常は治験責任医師が律義にこのメールを保管文書として打ち出してくれることは皆無であるため、CcにCRCを入れて協力を仰ぐ方が確実である。
Follow Up Letter
Follow Up Letterは、Confirmation Letterとは逆で、医療機関に訪問した後に、治験責任医師宛にCRAが送付するレターである。
Follow Up Letterには、積み残しの対応事項や、医療機関で協議した内容等、治験責任医師に情報共有しておくべき内容を記載するのが標準的である。
なお、Confirmation Letter、Follow Up Letterについては、GCPで規定された対応ではない為、運用をしていないプロジェクトもある。
その他の押さえておきたい用語

現場でのやり取りでの出現頻度はそれ程ないものの、用語としては知っておいた方が良いと思われる用語をまとめてみました。
現場での出現頻度が低いというだけで、重要度が低い訳では無いのでご注意を!
CSR
CSRとは、Clinical Study Reportの略で、治験総括報告書のことを指す。
治験総括報告書は、治験のデータを取りまとめた重要な資料で、いわば治験の集大成的な位置付けの資料である。
主に依頼者側にいなければあまりお目にかかることは無いが、CROでもCSR作成業務を受託していることがあるため、その際には目にすることがあるかもしれない。
なお、CRCの場合は恐らくCSRを見ることはほぼ無いと思われる。
CTN
CTNとは、Clinical Trial Notificationの略で、治験届のことを指す。
治験届には、依頼者、CRO、SMO、IRB、そして医療機関の情報等、治験に関連した様々な情報が記載されている。
この治験届は、初回に届け出た情報から更新がある際には、都度更新が必要であり、CRAは治験届に関連するような情報(医療機関名や住所、治験分担医師の情報等)に更新が無いかを一定期間ごとに確認することになる。
治験届に関連する情報の更新を見逃していた場合、薬機法に関わる部分のため、重めのミスとして認識されているので、特に注意が必要である。
ICH

ICHとは、International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Useの略であり、医薬品規制調和国際会議のことを指す。
日本語名で呼ぶことはほとんどなく、ほとんどの人が「ICH」と呼んでいる。
ICHは、グローバル化する医薬品開発に対応するため、世界的に統一した目線で医薬品の有効性・安全性・申請条件等を整備するために、日・米・欧の3極が発足した組織である。
医薬品開発における最新トレンドは、このICHで議論されていることがベースとなるため、医薬品開発に携わる身としては、最新の動向を押さえておきたいところだ。
今回の記事では、ICHをまだほとんど読んだことがなく、どこから手を付けたら良いか分からない方向けに、簡単な概要をまとめてみました。 また、2021年7月7日(水)に第43回ICH即時報告会がありましたので、その内容についても簡単に触れています。 “ICHは難しそう”と苦手意識を持っている方や新人の方、またこれから臨床開発の世界で働く予定の方の一助となれば幸いです!

ePRO
ePROとは、electric Patient Reported Outcomeの略であり、電子的な患者アウトカムのことを指す。
「電子的な患者アウトカム」と言ってもよく分からないと思うので、もう少し噛み砕くと、例えば、ある治験で、被験者の日々の症状について、被験者日誌で情報を収集するよう規定されていたとする。
被験者日誌は、紙でも良いのだが、例えば、スマートフォンなどでアプリをダウンロードし、そのアプリ上で日々の症状について入力をし、送信することがあるが、このシステムのことをePROと呼ぶ。
被験者が電子的に入力し、送信されたデータは、EDCに蓄積され、治験のデータとして収集される仕組みである。
この流れでは、転記が発生していないため、通常はSDVの対象外である。
まとめ
今回は、治験の現場において、実務で比較的よく出てくる用語についてまとめていきました。
本当は細かいことを言えば、もっと色々な用語があるのですが、あまりにも文字数が多すぎたので、重要かつ比較的出現頻度が高いものに絞っています。
この記事の用語集については、簡単な豆知識的な要素もいれてあるので、実務で専門用語が多すぎて困っている新人CRC、新人CRAの皆さんの一助になれば嬉しいです。
今後も、必要に応じて用語を追加していきたいと思います。






“治験に登場する用語について解説付きで紹介してみた” への1件のフィードバック