

令和2年8月31日に医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律が改正となり、それに伴いGCP省令も改正されました。
改正されたGCP省令には新たに「治験使用薬」という文言の定義が加わりましたが、GCP改正直後ということもあり、治験使用薬が新たに定義された経緯やそれがどのような薬剤を指すのかが今一ピンとこないという方も多いのではないでしょうか。
今回は、そんな治験使用薬について、私が勉強会や各種通知を通して確認した内容をまとめていきたいと思います。
はじめに
本記事を書いている2020年11月14日現在、治験使用薬に関する大まかな情報は、勉強会での説明や通知での記載で確認出来る状況です。
しかし、詳細な情報については今後QAにまとめて公開予定であるという方針を厚生労働省発表の勉強会で確認しています。
そのため、本ブログ投稿後に公開される予定であるQAの内容については随時更新をしていきたいと思っています。
本ブログは、【2020年11月14日現在】で私が知り得る情報をまとめています。
主に治験の実務的な部分で必要な情報について触れていきますが、初期段階であり私の認識が誤っている可能性もあるため、もし間違いが御座いましたらご指摘いただけますと大変助かります!
治験使用薬が定義された背景
今回のGCP改正で新たに「治験使用薬」というものが定義された訳ですが、そもそも何故新しい(しかも少々分かりにくい…)用語を定義する必要があったのでしょうか。
まずはその辺りからまとめていきたいと思います。
治験使用薬という新たな定義が加わった背景は、2018年5月9日に実施された医薬品医療機器制度部会での議論に遡ります。(私が知り得る範囲では恐らくこの時が起点かと思います)
この部会では、2017年7月6日に米医学誌「The New England Journal of Medicine」に投稿されたFDAの医薬品評価研究センター長による論文で治験の効率化の例としてマスタープロトコルが紹介されていることを取り上げ、日本の手続き上の問題について議論がされました。
まずは、マスタープロトコルについて簡単に紹介をしていきます。
マスタープロトコルとは?
最近では、がん領域において次世代シーケンサー(NGS:Next Generation Sequencer)の発達により、一部のがん種で遺伝子変異を標的にした治療法が確立してきています。
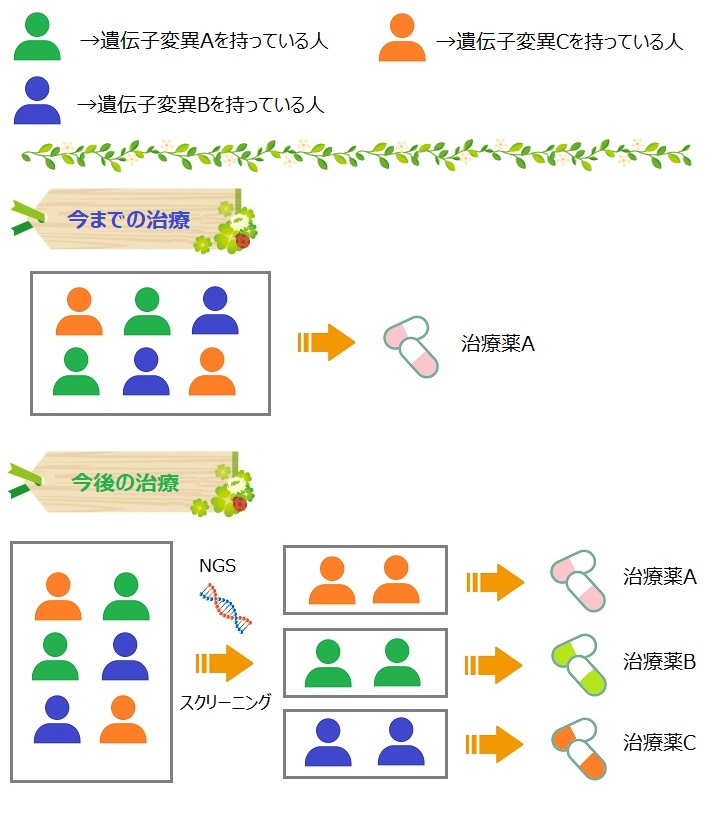
今までは特定の疾患について、治療薬が使用されていましたが、最近では遺伝子変異を特定し、分子サブタイプ別に治療薬(分子標的薬)が開発されてきています。
このような分子サブタイプ別に作用するような薬剤を開発する場合、治療薬A~Cでそれぞれ独立して第I相~第III相の治験を行うこともできますが、それぞれの試験を別々にやってしまった場合、統計解析計画書(SAP)やEDCなどの各種プラットフォームの仕様が試験により異なり、試験の結果を併合解析する際に不都合(異質性への対処)が生じてきてしまいます。
その問題を解決するのがマスタープロトコルになります。
マスタープロトコルは、アンブレラ型、バスケット型、プラットフォーム型の3つに分類することが出来るので、それぞれについて簡単に紹介をしていきます。
アンブレラ型マスタープロトコル
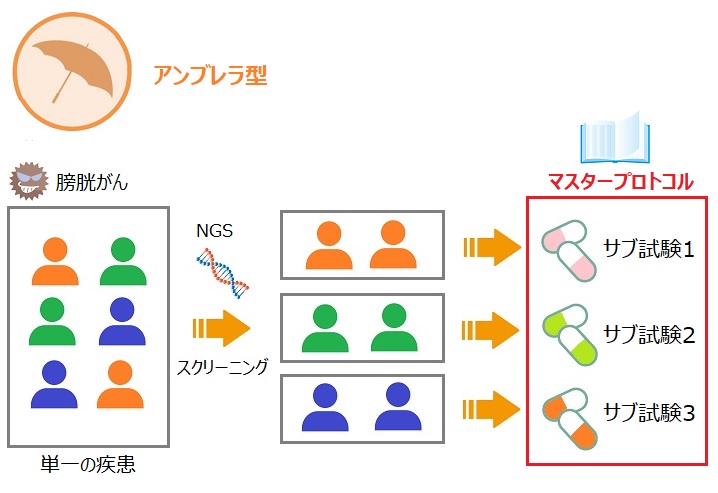
アンブレラ型マスタープロトコルは、単一の疾患に対して複数の治療法を対象とした試験に使用されるプロトコルになります。
アンブレラ型マスタープロトコルの試験では、上の絵のように単一の疾患(例では膀胱がんとしています)について、スクリーニングを行い遺伝子変異別にグループ分けをし、それぞれのグループに対して別々の治験薬を割り付けていきます。
しかし、ここで少し問題が…!
そうです、治験届です。
治験届は、原則1治験薬について1通の治験届を提出する必要があります。
つまりこの試験の場合は、3つの治験届を出す必要が出てきてしまうということです。
治験薬は3種類あるものの、マスタープロトコルはそれぞれのサブ試験共通のプロトコルになりますので、あくまで1試験です。
1試験なのに3つの治験届を出さなくてはいけないこの状況について、「非効率的だよね」ということを医薬品医療機器制度部会で議論していたということです。
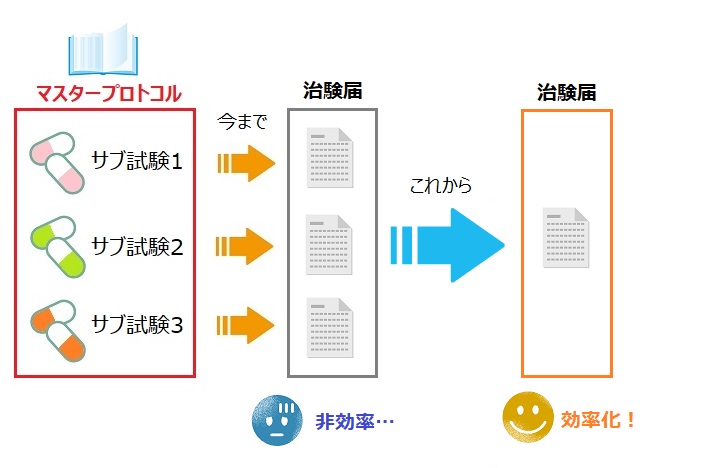
被験者群を“遺伝子変異別に分ける”という手法は最近までは無かったので、「1試験=1治験届」が成立していましたが、マスタープロトコルの登場で、「1試験≠1治験届」という状況も出てきており、今までの考え方では通用しなくなってきたのですね。
バスケット型マスタープロトコル
バスケット型マスタープロトコルは、複数の疾患に対して単一の治療法を用いる方法になります。
こちらも絵を使いながら見ていただいた方がイメージが沸くと思いますので、まずは下の絵をご覧ください。
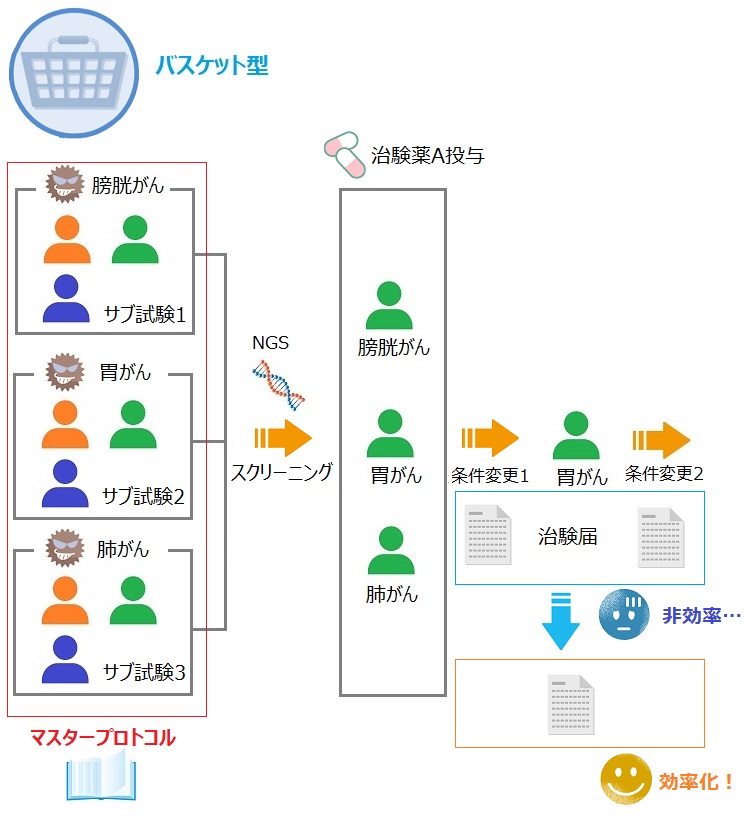
この例では、複数の疾患(膀胱がん、胃がん、肺がん)の中からある遺伝子変異を持つ被験者をスクリーニングして抽出し、そのそれぞれの被験者に対して単一の治療法(治験薬Aの投与)を行っています。
この時、膀胱がん、胃がん、肺がんの患者に治験薬を投与しているので、それぞれをサブ試験1~3とすることが出来ます。
アンブレラ型と同様にそれぞれについて治験をやることで色々と問題が出てくることから、このサブ試験共通のプロトコルとしてマスタープロトコルを使う試験がバスケット型になります。
先ほどのアンブレラ型と異なり、この例に示しているバスケット型の試験は治験薬を1種類しか使用していなので、バスケット型はもしかして治験届も1通で良いのでは?と思いますが、実はそうもいかないのです。
確かに治験薬は1種類なのですが、バスケット型の特徴として、投与計画を変更したり試験の目的を追加したりと大きな変更を加えていくことが挙げられます。
そして、この大きな変更を伴う場合は、治験届を新たに提出する必要が出てきてしまうのです。
そのため、やはりバスケット型においても1つの試験であるにも関わらず、複数の治験届を出さなければいけない状況となってしまうことがあるので、その部分について効率化が出来ないかという議論がされました。
具体的には、新たに治験届を出し直すのではなく、治験計画変更届で対応出来ないかといった検討がされました。
プラットフォーム型マスタープロトコル
アンブレラ型とバスケット型について触れていきましたが、実はもう1つプラットフォーム型マスタープロトコルというのがあります。
プラットフォーム型は、アンブレラ型とバスケット型を組み合わせたようなデザインなのですが、今回は、治験使用薬のお話がメインであり、アンブレラ型とバスケット型のデザインをある程度把握していれば問題無いので、詳しくは割愛させていただきます。
薬機法改正で変わった治験届の考え方
上記のように、マスタープロトコルの出現により治験届について運用を考え直す必要が出てきたため、今回の薬機法改正でルールが変わった訳ですが、具体的には以下のような考え方に変更となりました。
●原則として、被験者に生じた1件の有害事象につき、1件の副作用等報告とする。
●治験において用いた被験薬・対照薬・併用薬・レスキュー薬等の副作用は当局への報告を必要とする。
これだけでは「むむ?」となるかと思うので、もう少し詳しく見ていきたいと思います。
1つのプロトコルについて1通の治験届とする
こちらについては、先ほどのマスタープロトコルのところでも説明した通りになります。
アンブレラ型、バスケット型では、複数の治験届を出さなければいけない状態であったので、1通の治験届で対応できるように変更をしたということです。
被験者に生じた1件の有害事象につき、1件の副作用等報告とする
マスタープロトコルを使用した場合には、中身が複数のサブ試験で構成されていることから、1症例1件の有害事象(当局報告が必要な副作用)に対して重複して副作用報告がされる場合があります。
重複して副作用報告がされた場合には、複数の試験で副作用が発生したように見えてしまい、勘違いが発生してしまう可能性もあります。
そのため、1試験1治験届にすることで、副作用報告が重複してしまうことを防げるということのようです。
治験において用いた被験薬・対照薬・併用薬・レスキュー薬等の副作用は当局への報告を必要とする
ここで記載した「治験において用いた被験薬・対照薬・併用薬・レスキュー薬等」というのは、いわゆる今回定義された治験使用薬のことです。(治験使用薬の説明は後からしっかりするため、とりあえずここでは流しておいて下さい。)
これを聞いた時には私は「え…!?」となりました。併用薬についても副作用情報が必要なんて言ったら物凄いことになりそうで…
実は少しからくり?があるので説明を進めていきます。
治験のデザインによっては、治験薬(被験薬、対照薬)に加え併用薬を一緒に服用するデザインのものがあります。
このとき、治験薬によって生じた有害事象については、当局報告の対象となる条件が規定されていて、当局報告をしていたかと思いますが、併用薬については当局への報告は任意でした(よっぽど大きなことがあれば自発的に報告をしていたと思いますが…)。
被験者保護の観点から考えると今まで任意であった報告を必須とする方が妥当だろうという議論がなされ、副作用報告の対象に加わりました。
ここで重要なことは、治験実施計画書で規定された併用薬が対象ということ。
いつも収集しているあの併用薬全てについて副作用報告が必要ということではありません。
先ほどの絵で示したように特定の併用薬を使用することを規定した治験実施計画書の場合、その併用薬については副作用報告対象となるということです。
ここでまた1つ疑問が…
通常、このような場合は、併用薬が既に市販されているものであれば、市販後調査で副作用が収集されており、治験としては収集していなかったと思います。
そうです…今回の薬機法改訂により、市販後調査での副作用報告+治験での副作用報告が必要となってしまいました。
これについては、今後治験側での副作用報告に一本化できるように整備をしていく方針であるということですが、整備がされるまでは踏ん張っていかなければいけなそうな状況です…
先ほどは、治験薬+併用薬についてお話しましたが、実際にはレスキュー薬や前投与薬や併用機器等についても使用が規定されている治験実施計画書があると思います。
治験実施計画書で規定されているそれらについても副作用報告(機器の場合は不具合報告)の対象として加わりました。
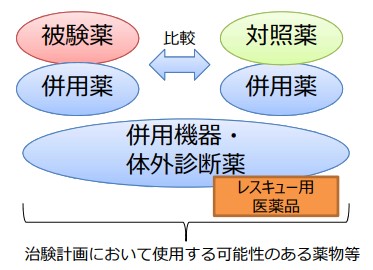
医薬品の治験で併用機器があった場合には、機器に詳しくない医師が不具合の評価をしなければいけないシチュエーションも想定できるため、医薬品メーカーも医療機器メーカーとの協力が必要となる場面が出てきそうです。
被験薬と治験使用薬の定義
さて、前振りはここまででいよいよ本題に入っていきたいと思います。
今回は治験使用薬についてのお話がメインなのですが、併せて被験薬の定義についても一緒に見ておいた方が良いので被験薬と治験使用薬についてそれぞれ説明をしていきたいと思います。
被験薬の定義
被験薬の定義については、今までと変わらないのですが、「治験の依頼をしようとする者による薬物に係る治験の計画の届出等に関する取扱いについて」に少し面白い記載があるので見てみましょう。
被験薬とは、治験の対象とされる薬物であり、当該治験の試験成績をもって当該薬物の製造販売承認申請を目的とするものを指す。
主たる被験薬とは、治験計画届出時に被験薬が1つの場合にはその被験薬を指し、複数の被験薬がある場合には、治験依頼者が選択した1つの被験薬を指す。
また、当該治験の試験成績をもって製造販売承認申請を目的とする医療機器(以下「被験機器相当」という。)及び再生医療等製品(以下「被験製品相当」という。)は、本通知の「被験薬」と同様の取扱いとすること。
上記の文章を見てお気付きですね?
そうです、「主たる被験薬」なるものが定義されているのです。
アンブレラ型のマスタープロトコルの絵を使って説明をしていきましょう。
アンブレラ型では、異なる治験薬を使用するため、それぞれに治験届が必要となってしまう仕様でした。
そこで、1つの治験届でOKとするよう、治験薬A~治験薬Cのうち治験依頼者が任意で「主たる被験薬」を選択し、決める事となりました。
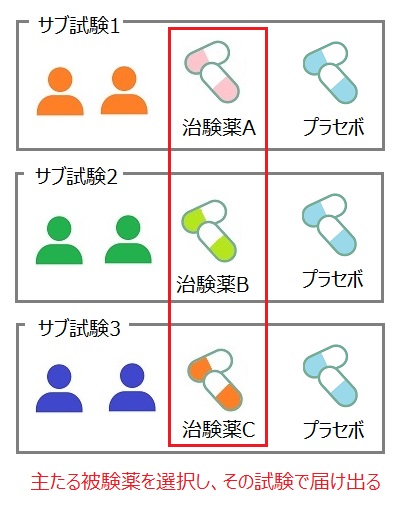
主たる被験薬は事務手続き上必要となるものであり、それ自体に大きな意味はありません。
その為、主たる被験薬を選ぶ当局から示す基準などは無く、任意に治験依頼者が決めて良いものになります。
これが治験届1本化の仕組みのうちの1つとも言えるでしょう。
治験使用薬の定義
治験使用薬の定義については、GCP第2条課長通知4の記載を見ていきましょう。
「治験使用薬」とは、治験において被験薬の有効性及び安全性の評価のために使用する、治験計画届書及び治験実施計画書において規定された既承認有効成分又は未承認有効成分を含む薬物(被験薬を含む。)を意味する。具体的には、被験薬、対照薬、併用薬、レスキュー薬、前投与薬等が該当する。
なお、GCP省令において「治験使用薬」に対して求める事項については、薬物に係る治験において被験薬の有効性及び安全性の評価のために使用する機械器具又は加工細胞に対しても、同様に遵守されることが望ましい。
出てきましたね、治験使用薬。
定義を読んでいただいて分かったかと思いますが、これは先ほど「治験計画において使用する可能性のある薬物等」にあったもの全てになります。
ただし、重要なのは、被験薬の有効性及び安全性の評価のために使用する、治験計画届書及び治験実施計画書において規定されたものであるということ。
つまり、プロトコルで規定されていないような併用薬については、治験使用薬には該当しないことになります。
逆に言えば、プロトコルで治験薬と一緒に飲むような併用薬が規定されていたり、全治療薬が規定されている場合には治験使用薬に該当する可能性(被験薬の有効性及び安全性の評価のために使用する場合)がありますので、注意が必要です。
また、先ほど少しお話した通り、今まではこの絵でいう被験薬と対照薬(つまり治験薬)については、副作用報告の対象となっていましたが、それ以外のものについては、副作用報告の対象とはなっていませんでした。
ここを義務化するために、一括りに治験使用薬として定義をし、従来までの治験薬と同等の重み付けをしたものと考えられます。(これは私見です)
治験使用薬の英訳
治験使用薬の英語表記については、2021年1月10日現在ではTwitter内で議論をしたところ、治験使用薬の定義的な意味から「drugs used in (the) clinical trial」が意味合い的にもシンプルで適切では?という結論に至りました。
なお、解析目線の意見では、治験使用薬はプロトコル毎にどのようなものを含めるか(レスキュー薬や前治療薬などがあるか等…)等があるため、目的に応じて英訳を使い分けるようになるのでは?という意見もありました。
その後、2022年2月22日に実施された製薬協による改正医薬品医療機器等法説明会 にて治験使用薬の英訳は、“Drugs used in the Clinical Trial”を推奨すると発表されてたことから業界ではこの表現が使用されることになるかと思います。

出典:改正医薬品医療機器等法説明会(令和4年2月22日)
治験使用薬が登場した初期段階からTwitter内でも色々な立場の方々とディスカッション出来たことに改めて感謝いたします!
治験届・副作用等報告の適用時期
新形式での報告には2年間の経過措置期間が設けられています。
つまり、令和2年9月1日から今回の薬機法改正が適用されたので、令和4年9月1日までが経過措置期間となります。
もっと噛み砕くと、この記事を書いている令和2年11月現在は、経過措置期間が始まったばかりで、恐らくほぼすべての治験が旧形式の副作用報告を継続している(つまり今までと同じやり方)と考えられるため、CRAやCRCさんの業務もそれほど影響を受けていないと思われますが、今後徐々に新形式に切り替わっていくというイメージです。
適用期間については文字だと分かりにくいので、以下の絵を見てもらう方が分かりやすいかと思います。
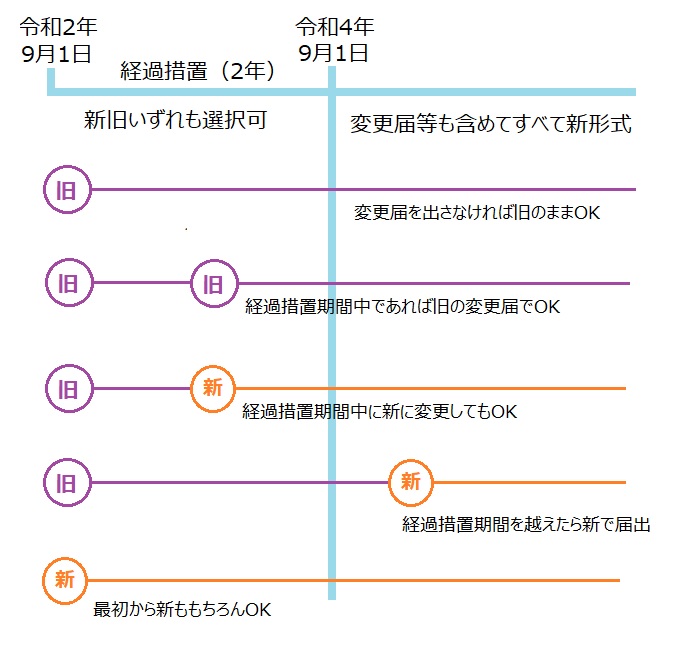
上記の絵で「新形式での報告」と記載されているものは、治験使用薬などの概念を盛り込んだ治験が実施されていることになります。
これから新規に治験が立ち上げる場合、令和4年9月1日までに治験終了届が出せるのであれば、経過措置期間中に旧形式で治験計画届を提出して、今まで通りのやり方でその治験は完結できますが、令和4年9月1日を跨ぐような治験の場合には、予め新形式で治験を開始させるか、あるいは試験の途中で新形式に変更する対応になるかと思います。
これは個人的な考えですが、試験の途中で新様式にいきなり変わると現場は混乱するかと思うので、令和4年9月1日を跨ぐ可能性が高いのであれば、新規の治験は新形式でスタートした方が良いのではないかと思っています。(私のプロジェクトはその方向で進めることを検討しています…)
まだ経過措置期間が始まったばかりで実感は無いかもしれませんが、確実に切り替わるタイミングがやってきますので、今のうちからある程度どのような不都合が生じるのかなどを想定しておく必要があるかもしれません。
ということで、もっと実務的なお話に入っていきたいと思います。
治験使用薬の登場で色々と変わります…
治験使用薬の登場で変わること(主に現場で)
現時点では、GCP改訂に関するQ&Aがまだリリースされていませんが、今後、段階的にQ&Aをリリースしていくとのことです。
細かい部分で様々な問題が出てくるかと思いますが、現時点で私が把握している情報で気になる項目をピックアップして記載してみようと思います。
今後の展開次第では、対応が変更となる可能性も考えられるため、情報がアップデートされた際には、なるべく本記事についても情報をアップデートしていきたいと思います。(弱小個人ブログなので追いつくかは不明ですが…)
なお、今回のGCP改訂でどこが具体的に変わったのかについては、ポケット資料集の見え消し版が非常に役に立ちます。
それでは見ていきましょう。
初回審議資料に「治験使用薬(被験薬を除く。)に係る科学的知見を記載した文書」が追加された
今までは、ご存知の通り、GCP第10条で治験薬概要書の提出が規定されていましたが、今後は治験薬概要書に加えて、「治験使用薬(被験薬を除く。)に係る科学的知見を記載した文書」も追加で提出する必要が出てきます。
そして、「科学的知見を記載した文書」とは…?具体的になに…?となる訳です。
治験使用薬に未承認治験薬が使用されている場合には、最新の治験薬概要書を用意することが望ましいようですが、実際には他社の場合難しいことも多々あると思います。
その場合は、安全性が担保できる場合に限り、インタビューフォーム、添付文書や学術論文等の公的にリリースされている資料を添付することでも良いとのことです。
海外の安全性情報については、日本と同等の薬事がなされている国の情報であれば使用可能と考えるとのことですが、まだ少し曖昧な気もしますね…
今後Q&Aでより詳細に詰めていくのでしょうが、とりあえず初回審議資料が増える事には変わりなさそうです(と言っても、治験使用薬が実薬とプラセボのみの場合であれば今までと変わらないのですが)。
治験薬の管理に治験使用薬の管理が加わった
GCP第16条で、記録・管理に関する手順書に関する管理すべき対象に治験使用薬が加わりました。
今までは、治験薬管理手順書に従い、治験薬を取り扱っていたかと思います。数量管理や温度管理の記録も取っていたかと思います。
ここに治験使用薬が加わってしまいました。
しかし、GCP第39条の課長通知4を見てみると以下のようなことが書いてあります。
実施医療機関の長又は治験薬管理者は、治験依頼者が作成した治験使用薬の取扱い及び保管、管理並びにそれらの記録に際して従うべき指示を記載した手順書(第16条第6項参照)に従い、実施医療機関に交付された治験使用薬の受領、実施医療機関での在庫、被験者ごとの使用状況及び未使用治験使用薬の治験依頼者への返却又はそれに代わる処分に関して、記録を作成し、保存すること。
これらの記録には、日付、数量、製造番号又は製造記号、使用期限(必要な場合)並びに治験薬及び被験者識別コードを含むこと。
また、治験薬以外の治験依頼者が交付しない治験使用薬であって、実施医療機関が在庫として保管するものの中から使用する治験使用薬については、治験依頼者は、実施医療機関において定められた取扱い、保管、管理、処方等に係る手順等に基づき対応すること。
治験使用薬についても、温度管理記録や数量管理記録が求められたら物凄く大変になるなと思っていましたが、対象は依頼者から施設へ交付したものに限定されるようです。
治験薬については、当然依頼者から施設に交付することになるので、今まで通りの記録が必要として、プロトコルで規定された併用薬やレスキュー薬等がある場合は、依頼者から提供したものかそうでないかで対応が分かれることになります。
今まで依頼者に併用薬やレスキュー薬の搬入を要望していた施設は、治験使用薬についての管理記録各種の対応が必要となるのでCRA、CRCさん共に負荷が増す可能性があります。
施設の在庫の医薬品を使用する場合には、施設のやり方で管理することでOKとなっているので、個人的には施設の在庫を使用する方がお互いにとって良いのかもしれません。
有害事象の定義が変わった
GCP第2条の課長通知13に記載されていた有害事象の定義が以下のようになりました。
「有害事象」とは、治験使用薬又は製造販売後臨床試験使用薬を投与された被験者に生じた全ての好ましくない又は意図しない疾病又はその徴候(臨床検査値の異常を含む。)をいい、当該治験使用薬又は当該製造販売後臨床試験使用薬との因果関係の有無は問わない。
もしかすると、この対応が一番厄介と言っても過言ではないかもしれません。
特に治験使用薬が複数ある場合、その治験使用薬毎に因果関係を調査する必要が出てきます。
今までは、「治験薬との因果関係」で考えていたものを「治験使用薬との因果関係」と考えることで、いきなり収集しなければいけない情報の範囲が広がったイメージです。
また、先ほども少し触れましたが、プロトコルで規定された治験使用機器相当があった場合は、機器との因果関係等も見なければいけないので、先生もその判断に迷われることがあるかと思います。
その為、製薬メーカーも医療機器メーカーと協力する必要があるというお話でしたが、果たして本当に大丈夫なのか不安なところではあります…
なお、正確にはGCPには併用する医療機器も治験使用薬と同様に扱う事が望ましいと記載されているので、必須ではないのかもしれませんが、その辺りはどうなのでしょうね。
蛇足ですが…
サラッと、「治験使用機器相当」と使いましたが、これはどうやら「治験使用機器」にしてしまうと、その治験が、医薬品の治験なのか医療機器の治験なのか分からなくなってしまうため、医薬品の治験で併用機器がある場合は、「治験使用機器相当」と便宜上使うようです。
GCPには「治験使用機器相当」という言葉は出てきませんが、「「治験の依頼をしようとする者による薬物に係る治験の計画の届出等に関する取扱いについて」には出てきますよ!
なので、医療機器の治験で併用薬があるときには、逆に「治験使用薬相当」と呼ばれることになります。
令和4年8月31日には薬機法の経過措置期間が終了し、9月1日からは完全移行となります。しかし、現時点では導入の実例報告も少なく、9月1日の完全移行を前にして業界内でも困惑の声を聞く機会があります。 そこで、早期段階より治 …

まとめ
細かいところを詰めれば、もっと気になるところなどはたくさんあるのですが、情報があまり出ていない現段階においては、概ねこの記事で紹介した内容を知っておけば良いのかなと思っています。(治験使用薬と関係ないところでは、治験情報についてjRCTに1本化されたという変更等もあります)
今後、段階的にQ&Aがリリースされていくとのことですので、そちらでも補足説明がされるかと思いますし、更に12月にはGCPリノベーションセミナーもあるので、そちらでもお話があるかもしれません。
また今回は、自分の勉強の為にも一度把握している情報を整理する意味でこの記事をまとめたのですが、この記事の一部分だけでも皆さんの知識の糧になればとても嬉しいです。
なお、本記事は、治験使用薬追加の背景にスポットを当ててお話をしていきましたが、実際の現場でどのような影響があるのかについては以下の記事にまとめてありますので、併せてご覧いただけますと幸いです。
令和2年8月31日に薬機法が改正され、それに伴いGCP省令も改正されました。 前回は、新たに加わった治験使用薬の追加背景や定義について詳細に触れていきましたが、その後、Q&Aなどが更新され、その運用がより具体的になってきました。 今回の記事では、治験使用薬が加わることで主に現場で留意しておく必要がありそうなポイントや今までの運用とどのように変わっていったのかを紹介していきたいと思います。 加えて、令和3年8月施行分の内容についても触れていきますので予め予習をしておくのにもお使いいただければ幸いです。







“GCP改正で追加された治験使用薬とは?背景を含めて解説” への2件のフィードバック