

令和2年8月31日に薬機法が改正され、それに伴いGCP省令も改正されました。
前回は、新たに加わった治験使用薬の追加背景や定義について詳細に触れていきましたが、その後、Q&Aなどが更新され、その運用がより具体的になってきました。
今回の記事では、治験使用薬が加わることで主に現場で留意しておく必要がありそうなポイントや今までの運用とどのように変わっていったのかを紹介していきたいと思います。
加えて、令和3年8月施行分の内容についても触れていきますので予め予習をしておくのにもお使いいただければ幸いです。
現在の状況を整理
 実のところ、この手のお話は法律などがごちゃごちゃしており、非常に分かりにくい状況になっています。そのため、まずは簡単に今は一体どのような状況になっているのかをお話します。
実のところ、この手のお話は法律などがごちゃごちゃしており、非常に分かりにくい状況になっています。そのため、まずは簡単に今は一体どのような状況になっているのかをお話します。
令和2年8月31日に薬機法が改正されましたが、それに伴い、医薬品の臨床試験の実施の基準に関する省令(つまり、GCP省令ですね)のガイダンスも同日に改正され通知されました(薬生薬審発0831第15号)。
ここで追加されたのが、「治験使用薬」でした。
治験使用薬の追加の背景などについては、前回の記事にまとめてありますので、まだご覧になっていない方は是非そちらもご覧下さい。
令和2年8月31日に医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律が改正となり、それに伴いGCP省令も改正されました。 改正されたGCP省令には新たに「治験使用薬」という文言の定義が加わりましたが、GCP改正直後ということもあり、治験使用薬が新たに定義された経緯やそれがどのような薬剤を指すのかが今一ピンとこないという方も多いのではないでしょうか。 今回は、そんな治験使用薬について、私が勉強会や各種通知を通して確認した内容をまとめていきたいと思います。

まずは、ここまでの状況を簡単に図にしてみますと以下のようになります。
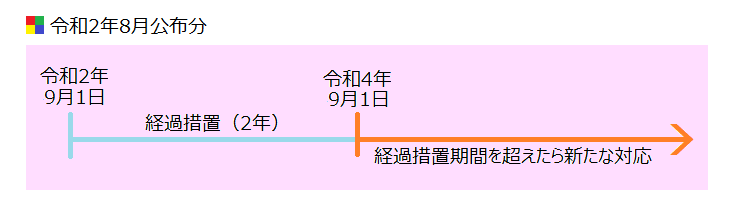
完全移行が、令和4年9月1日ですので、現時点(令和3年4月)からは1年以上先になりますが、各方面(医療機関や依頼者等)でSOPの整備が進められているため、実際には完全移行の前から運用が始まることが想定できるため、早めに知識を詰め込んでおく必要があるかと思います。
そして、令和2年8月31日(令和2年厚生労働省令第155号)に続き、令和3年1月29日(令和3年厚生労働省令第15号)に更に薬機法が改正されました。先ほどの改正分と合わせて見てみると以下のようになります。
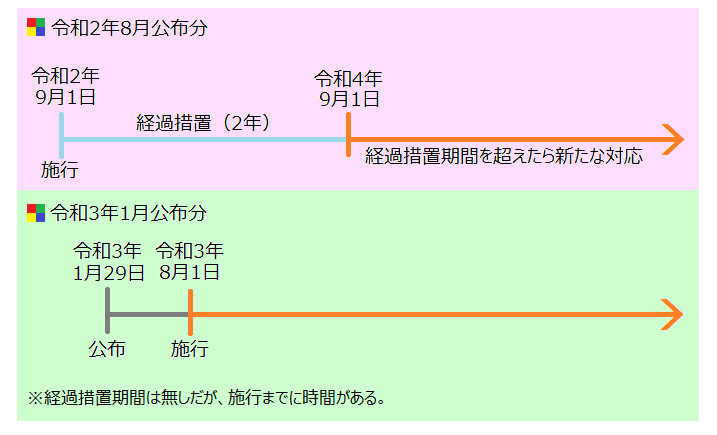 令和3年1月公布分、令和3年8月施行の内容であるため、まだGCPガイダンスは出ていないものと思われますが、今後GCPガイダンスも出てくるかと思うので、予めどのような変更があるのかを見ておくことをおすすめします(詳しくは後述します)。
令和3年1月公布分、令和3年8月施行の内容であるため、まだGCPガイダンスは出ていないものと思われますが、今後GCPガイダンスも出てくるかと思うので、予めどのような変更があるのかを見ておくことをおすすめします(詳しくは後述します)。
上記は、法律やガイダンスについてのお話でしたが、その他、「E2B(R3)実装ガイドに対応した市販後副作用等報告及び治験副作用等報告に関するQ&Aの改正について」も令和2年12月9日に出ており、治験使用薬について明記されたため、今回の記事ではそちらの内容も踏まえてお話をしていきます。
治験使用薬関連
前回と重複する内容もありますが、治験使用薬関連についてまとめていきます。主には現場で重要となるポイントに焦点を当てつつ、議論が必要な個所や注意すべき個所については具体的な例も挙げていきたいと思います。
用語周りの整理
治験使用薬の登場に付随して、「治験の依頼をしようとする者による薬物に係る治験の計画の届出等に関する取扱いについて」の資料で、今までに無かった用語が定義されています。
●被験機器相当/被験製品相当
●治験使用機器相当/治験使用製品相当
こちらについては、以前の記事でも触れましたが、簡単に触れておきます。それぞれの用語については、以下のようなイメージになります。
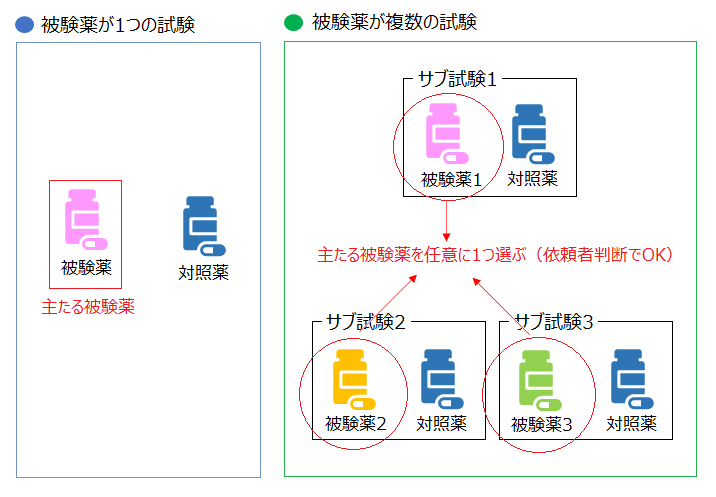
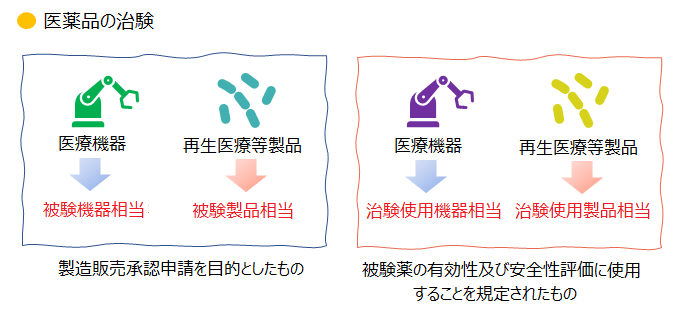
用語をご覧いただくと、「○○相当」というような表記があるかと思います。
これは、手続き上の問題で、その治験が、医薬品の治験なのかそれとも医療機器の治験なのかを一目で明確に区別するために設定された背景があります。
そのため、医療機器の治験の場合は、上記の例とは変わり、「治験使用薬相当」という言葉出てきます(詳細は「機械器具等に係る治験の計画等の届出の取扱い等について」を参照)。
このことは、主に届出上のお話のため、CRAやCRCさんにはそれほど関連が深い内容では無いかもしれませんが、何かのタイミングで話題になることもあるかもしれませんので、頭の片隅にでも置いておいていただくと良いかもしれません。
有害事象の定義が変更
治験使用薬の登場によって1番大きな影響を受ける項目が、この「有害事象の定義の変更」ではないでしょうか。
有害事象の定義が変更になったことで、副作用等報告にも影響が及ぶため、CRAやCRCさんのみならず非常に広い範囲の部門の対応フローが変わってくるものと思われます。
新たな定義は以下のようになりました。
「有害事象」とは、治験使用薬又は製造販売後臨床試験使用薬を投与された被験者に生じた全ての好ましくない又は意図しない疾病又はその徴候(臨床検査値の異常を含む。)をいい、当該治験使用薬又は当該製造販売後臨床試験使用薬との因果関係の有無は問わない。
文言だけを見ると、「治験薬」⇒「治験使用薬」のように少し言葉が変わっただけですが、これだけでも有害事象としてピックアップしなければいけない安全性情報が非常に広くなりました(後程図解で示します)。
上記に加え、見逃してはいけないのが以下の記載です。
「治験使用薬」とは、治験において被験薬の有効性及び安全性の評価のために使用する、治験計画届書及び治験実施計画書において規定された既承認有効成分又は未承認有効成分を含む薬物(被験薬を含む。)を意味する。具体的には、被験薬、対照薬、併用薬、レスキュー薬、前投与薬等が該当する。なお、GCP省令において「治験使用薬」に対して求める事項については、薬物に係る治験において被験薬の有効性及び安全性の評価のために使用する機械器具又は加工細胞に対しても、同様に遵守されることが望ましい。
つまり、併用機器等がある治験においては、その併用機器についても治験使用薬と同様に有害事象に対しての判断をすることが望ましいとされているのです(因果関係の判定など)。
これらを考慮すると、有害事象が発生した際には、以前と比較し、以下のように変更することになります。
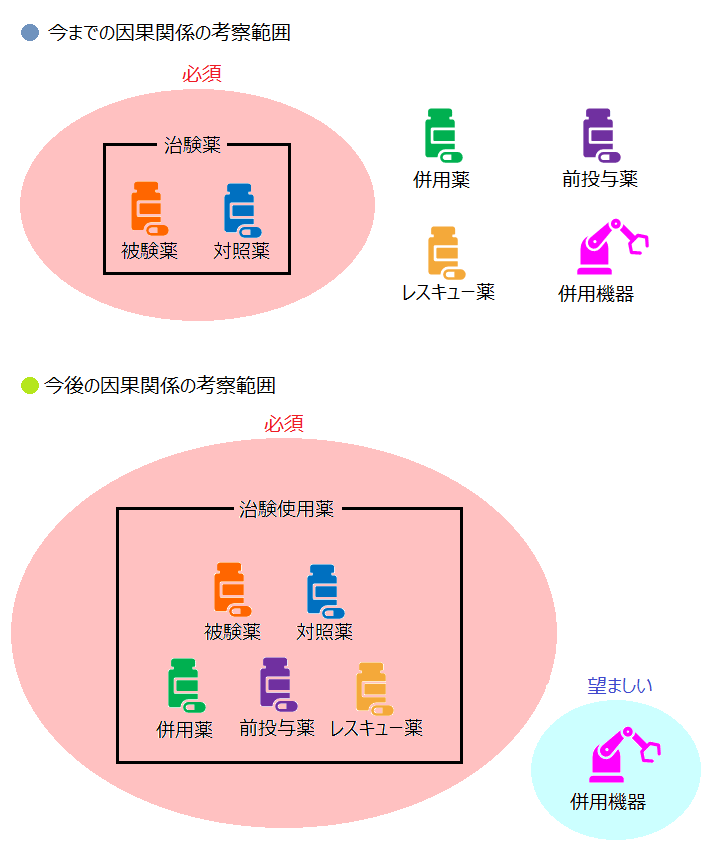
併用機器の安全性情報の収集は、「望ましい」とあるので、必須ではないのでしょうが、もし同様に有害事象の収集をするのであれば、確認をしなければいけない範囲が広がるばかりではなく、場合によっては医療機器についての安全性評価も医師が判断しなければいけないことになり、専門外の場合、適切に判断ができるのかという疑問も湧いてきます。
場合によっては、医療機器メーカー等との連携も必要になってくることが想定されますが、現場を見てきている身としては、あまり現実的ではない気がしています。
副作用情報等
「治験安全性最新報告概要」と「国内重篤副作用等症例の発現状況一覧等」を定期報告時に実施医療機関の長と治験責任医師に提供する対応は変わっていませんが、有害事象の定義が変更になったこととリンクして、報告対象が「治験薬」⇒「治験使用薬」になっています。
重篤な有害事象(SAE)が発生した際の対応フローについても、「治験薬」⇒「治験使用薬」となったのみで、国内治験においては、報告期限については今まで通りの対応となります。
| 予測性 | 重篤性 | 当局への報告期限 | |
| 被験薬 | 未知 | 死亡・死亡につながるおそれ | 7日 |
| その他重篤 | 15日 | ||
| 既知 | 死亡・死亡につながるおそれ | 15日 | |
| その他重篤 | 不要 | ||
| 被験薬以外の治験使用薬 | 未知 | 死亡・死亡につながるおそれ | 7日 |
| その他重篤 | 15日 | ||
| 既知 | 死亡・死亡につながるおそれ | 15日 | |
| その他重篤 | 不要 |
今までとやるべきことは同じなのですが、因果関係の部分のみ注意が必要です。
先ほども触れた通り、今までは、治験薬との因果関係が否定できないAEが副作用とされていましたが、今後は、治験使用薬との因果関係で判断しなければなりません。
つまり、今後は副作用として取り扱われる情報が増えることになり、結果として各医療機関への安全性情報の提供頻度の増加が想定されます。
そのため、現場では以下のような状況が想定されます。
●書式16(安全性情報)の発出頻度が増える。
●書式16の発出頻度が増えることによりIRBへの対応事項も増える。
●当局への安全性報告の頻度が増える(依頼者)。
上記からも分かる通り、やはり業務量としてはどうしても増えてしまいそうな印象を持っています(CRAもCRCさんも)。
また、上記に付随して、「モニタリング報告書/Contact Reportの作成頻度が増える」、「QCで確認しなければいけない資料が増える」、「Inhouse CRAの対応事項が増える」、「資料保管(TMFへの移管作業)が増える」など、幅広い業務に波及してしまうため、見た目以上にインパクトが強いことも想定できます。
場合によっては、CROコストが増加し、依頼者側の委託負担額が大きくなる可能性も大いにあり、私自身もどのようになるのか注視しています。
これは、私見ですが、グローバル試験から日本が排除されつつある現状の1つに”コストが高い”ということがあるかと思いますが、今回の一連の変化により業務が煩雑化し、更にコストが上がらないかどうかという点は非常に危惧しています。
治験計画届、IRBの審議資料の追加
治験使用薬が追加されたことにより、治験計画届に添付する資料と初回IRBの審議資料に、「治験使用薬に係る科学的知見を記載した文書」が必要となります。
表現が抽象的でやや分かりにくいのですが、「治験使用薬(被験薬を除く)に係る科学的知見を記載した文書」とは具体的に以下のような資料を指します。
●インタビューフォーム
●添付文書や学術論文等の公的にリリースされている資料
※GCP第10条ガイダンスを参照
この変更については、IRB審議資料にも関わる内容であることから、運用の初期の頃について、CRAは、IRB SOPがしっかりと更新されているかを確認する必要が出てきます。
多くのIRBで、SOPがしっかりと更新されているものと思いますが、万が一、SOPが更新されていないようでしたら、初回IRBまでに更新いただくようお願いした方が良いかと思います。
また、それでも間に合わない場合は、「その他、治験審査委員会の委員長が必要と認めた資料」に入れ込むのも良いかとは考えますが、なるべくなら早い段階でIRB SOPは確認しておきたいところです(ここはしっかりと事務局見解と依頼者見解の確認が必要な部分かと思います)。
更に、初回審議資料で提出した資料に更新があった場合には、基本的には、書式10(治験に関する変更申請書)を提出して審議が必要かと思いますので、治験使用薬の概要書や添付文書を提出した際には、更新時に注意が必要になります。
レスキュー薬や前投与薬については、他社製品を使用していることも多くあるため、添付文書の改訂があった際には感知が遅れる可能性もあり、オペレーションの立場としてどう管理していこうか私も模索しています。
治験使用薬の管理方法の変更
GCP第39条では、治験薬管理者について、治験薬の管理責任が記載されていましたが、治験使用薬の管理責任へと範囲が拡大しました。
依頼者側についても、GCP第16条が変更となり、「記録・管理に関する手順書」で管理すべき対象が、治験使用薬へと範囲が拡大しました。
治験使用薬の具体的な管理についてはGCP第39条のガイダンスに以下のように記載されています。
実施医療機関の長又は治験薬管理者は、治験依頼者が作成した治験使用薬の取扱い及び保管、管理並びにそれらの記録に際して従うべき指示を記載した手順書(第16条第6項参照)に従い、実施医療機関に交付された治験使用薬の受領、実施医療機関での在庫、被験者ごとの使用状況及び未使用治験使用薬の治験依頼者への返却又はそれに代わる処分に関して、記録を作成し、保存すること。
これらの記録には、日付、数量、製造番号又は製造記号、使用期限(必要な場合)並びに治験薬及び被験者識別コードを含むこと。また、治験薬以外の治験依頼者が交付しない治験使用薬であって、実施医療機関が在庫として保管するものの中から使用する治験使用薬については、治験依頼者は、実施医療機関において定められた取扱い、保管、管理、処方等に係る手順等に基づき対応すること。
また、治験実施計画書に規定された量の治験使用薬が被験者に投与され、治験使用薬の数量が正しく管理されたことを示す記録を作成し、保存すること。
今までの治験薬管理表の対象範囲が、「治験薬」から「治験使用薬」になることによって拡大したのですが、赤字部分で施設の在庫分の治験使用薬(治験薬を除く)を使用する場合は、医療機関の運用で管理して良いことが記載されています。
そのため、レスキュー薬や前投与薬などの治験使用薬が規定されている治験においては、依頼者から提供を受けるよりも、施設の薬を使用した方が施設側としても運用が楽になる気がしています。
ただ、請求という観点で考えると、依頼者から提供を受けた方が煩雑ではないかと思いますので、その辺りはどちらを取るのかというところでしょうか。
また、もし依頼者提供とした場合、依頼者の手順書で規定された要件を満たす管理表の種類が増えますので、CRCさんはもちろんのこと、CRAもDAをする範囲が広がり、今まで以上に時間が取られてしまうかもしれません(意外に管理表と在庫との整合性を見るの大変ですよね…)。
なお、医療機関で準備した治験使用薬(治験薬を除く)について、施設の運用で管理をして良いとなっていますが、治験119では製薬協が以下の見解を述べています。
該当するGCPガイダンスにも記載がありますように、治験薬以外の治験依頼者が交付しない治験使用薬であって、実施医療機関が在庫として保管するものの中から使用する治験使用薬については、基本的に実施医療機関において定められた取扱い、保管、管理、処方等に係る手順等に基づき対応することで差し支えありません。
ただし、適切に保管、管理された治験使用薬が、治験実施計画書に規定された量で被験者に投与されたことを示す記録を作成し、保存する必要があることから、どの被験者にどのくらいの数量の治験使用薬が使用されたかがわかる出納記録のような記録が必要と考えます。
ロット番号も含め、どのような情報を記録に残すべきかは、事前に治験依頼者と協議することをお勧めします。
上記からすると、「施設のやり方でOK」とGCPでは記載されているものの、結局は試験開始前に記録の残し方について協議することになりそうです。
もちろんここで依頼者の主張を押しすぎると…あとは、ご経験者の方ならどうなるか予測できることでしょう。。
その他運用関連

治験使用薬関連の変更が非常に大きなウェイトを占めているため、その他の変更については、治験使用薬程大きなインパクトはありませんが、中でも実務をしていく上で影響しそうな項目についてピックアップしていきます。
同意文書に記載すべき情報の変更
GCP第51条が変更となり、同意説明文書に記載すべき事項から分担医師に関する情報が削除されました。
治験使用薬程はインパクトが無いものの、ICFに分担医師の情報を記載しなくても良くなったことは大きいことかと思います。
特に規模が大きい医療機関の分担医師は、比較的異動が多いため、その都度、ICFの改訂が必要となると、非常に負荷が大きいです。
分担医師の情報が削除されたことにより、ICFの改訂頻度が下がることが期待できるため、責任医師、CRCさんを始め、CRAやその他のスタッフについても全体的に負荷が減ることが期待できます。
同意説明文書は、ご存知の通り、治験責任医師が作成する資料であるため、施設SOPにも同意説明文書関連の記載があるはずですので、CRAは施設SOPについても“分担医師の情報の記載が不要であること”をしっかりと確認しておきたいところですね(施設SOPに記載が残っていた場合は、勝手に分担医師の情報を削除したバージョンでFIXすると、SOP違反になってしまいます)。
CRCさんは把握されているので大丈夫かと思いますが、CRAで新人のうちは見落としがちになる箇所ですので、注意が必要かと思います。
症例報告書(CRF)関連の変更
さらっと見ていると、あまり気にせずスルーしてしまいそうな内容が多いのですが、症例報告書関連(第47条)についても変更・追記が比較的多くあります。
概要は以下の通りです。
①CRFの内容点検が、治験責任医師等から治験責任医師に限定された(分担医師は不可に)。
②電子データ処理システムに対してCRFのデータを入力することもできることが明記された。
大体の試験は、EDCを使用していると思うので、①についてはあまり影響が無いかと思います。最終的に責任医師が内容を確認していることについては、EDCの場合、治験責任医師の電子署名で対応しているので問題ありません。
②については、DDCの普及などで明記されることになったのでしょうかね。
上記以外にも②については、バリデーションされたものを使用することなどの規定が明記されましたが、さすがに当たり前の対応で今までも実施されている(はず)なので、影響はあまり無いかと考えています。
治験使用機器
治験使用機器関連については、以下のページにまとめてあります。
なかなか登場する場面は少ないかもしれませんが、医薬品の治験で治験使用機器を定義する場合に参考になればという思いで私の経験上のお話をさせていただいておりますので、よろしければ是非ご覧下さい!
令和4年8月31日には薬機法の経過措置期間が終了し、9月1日からは完全移行となります。しかし、現時点では導入の実例報告も少なく、9月1日の完全移行を前にして業界内でも困惑の声を聞く機会があります。 そこで、早期段階より治 …

令和3年1月公布分(8月施行)で変更となったこと

令和3年1月に公布された内容は、令和3年8月から施行になります。記事を執筆している時点(令和3年4月8日時点)では、GCP省令やGCPガイダンスの連絡は来ていませんので、お手元の2021年版のポケット資料集でも情報が反映されていないかと思います。
GCP省令やガイダンスについては、8月の施行までの間に情報が出るかと思いますので、参考程度にご覧下さい。
治験実施計画書に記載すべき事項が変わる
第七条(治験実施計画書)の項目で、第十五条の四第三号と第七号の表記が削除されました。
つまり、以下のように変わります。
②治験薬提供者の氏名及び住所が不要になった。※②は、医師主導治験のお話です。
CROの情報がプロトコールより無くなることで、情報更新があった際のプロトコール改訂(あるいは別冊改訂)が減るかと思いますが、そもそも頻回に変更することは少ないかと思いますので、業務量にもほとんど影響しないと思われます。
ただし、治験届には今まで通り、CROの情報が必要となりますので、注意が必要です。
治験薬に添付する文章や容器若しくは被包への記載ルールが変わる
今までは、「予定される販売名」、「予定される効能効果」、「予定される用法用量」について、拡大治験を実施する場合のみ、「この限りではない」と規定されていました。
その部分について、今回の改定では、拡大治験に加え、非盲検試験においても「この限りではない」の対象に含まれました。
治験薬のラベリング時点での影響はあるかもしれませんが、業務量が増えるというよりも減る印象ですので、ポジティブな改定なのではないでしょうか(あまり詳しくないので、隠れた影響もあるのかもしれませんが…)。
モニタリング報告書の記載ルールが変わる
これは、地味な改定なのですが、今までは「モニタリングを行った日時」を記載しなければいけませんでしたが、これからは、「モニタリングを行った日付」で良いことになりました。
これは、モニタリング報告書を書いている方は少し分かるかと思いますが、実際にモニタリングをしていた時間やその他の資料との整合性などを考えていると少々面倒な部分がありました。
さほど大きなインパクトではないかもしれませんが、CRAとしては地味に嬉しい改定かと思います。
同意説明文書に記載
先ほど、令和2年9月施行分として、「分担医師の情報が記載不要になった」ことを記載しましたが、令和3年8月施行分からは、更に「治験責任医師の職名」も記載不要になります。
つまり、同意説明文書に記載するのは、PI、SIの情報は、「治験責任医師の氏名及び連絡先」に変更となります。
令和3年8月から随分とサッパリしそうですね!
記載しなければいけない情報量が減るので、喜ばしい改定かと思います。
まとめ
今回は、令和2年9月施行分と令和3年8月施行分の内容についてお話をしていきました。
治験使用薬の追加を始め、非常に大幅な改定となるため、私たちの業務の仕方、考え方もこれから大きく変わってくることが想定されます。
また、現時点においては新たらしい運用でスタートしている治験はほぼ無い状況ですので、どのようなリスクが潜んでいるか未知数なのが正直なところかと思います。
個人的には、副作用の因果関係判定について、「治験薬との因果関係」から「治験使用薬との因果関係」に変更となったことから、生じた有害事象が「被験薬によるものなのか」あるいは「被験薬以外の治験使用薬によるものなのか」の判別がつきにくくなってしまうのではないかと心配しています。
更にそれが添付文章上にどう記載されるのかも気になります。
まだまだ未知数なところが多いのですが、今後も情報の更新がある度にブログも更新していこうと思います。





“GCP改訂ポイントと治験使用薬周りの運用まとめ” への1件のフィードバック