

CRAやCRCさんからしたら避けては通れない重篤な有害事象(SAE:Serious Adverse Event)の対応。SAE対応は時間制限があるため、いざ報告書を作成しようと思ってもじっくり考える機会も少ないかもしれません。
そこで、SAE報を受領した依頼者の立場から提出されるSAE報で気が付いた点などをまとめていきたいと思います。
はじめに

本記事は、新人の方も多く見ていただいているので、いつも通り基礎的な部分からお話をしていきたいと思います。
また、「SAE報告の本質的な意味を理解すること」を目標にしているため、新人研修などの内容よりも少し踏み込んで紹介をしていきます。
本質的な意味を理解することで、施設、SMO、CRO、依頼者との認識がうまくハーモナイズして円滑な治験運営に繋がって欲しいという願いを込めて書き綴っていきます。
なお、依頼者やプロジェクトによって微妙に状況が異なると思いますので、本記事は私視点で書かれたものである点にご留意下さい。
重篤な有害事象(SAE)
.jpg)
重篤な有害事象(SAE)は、有害事象のうち以下に該当するものです。
- 死に至るもの
- 生命を脅かすもの
- 治療のため入院または入院期間の延長が必要となるもの
- 永続的または顕著な障害・機能不全に陥るもの
- 先天異常をきたすものその他の医学的・科学的根拠に基づいて重大と判断されたもの
疾患領域にもよりますが、「入院または入院期間の延長」でSAEとなるケースが多く、症状が重い領域(オンコロジー領域など)は死亡のSAEの遭遇もあります。
また、「入院」に関しては例えば、「白内障の手術で日帰り入院をしたらSAEですか?」や「検査入院はSAEですか?」みたいな質問が出てきますが、治験前より予定されている入院についてはSAEとはしないことが一般的です。
このことは、SAE報告についての事務連絡「E2B(R3)実装ガイドに対応した市販後副作用等報告及び治験副作用等報告に関する Q&A について」のQ10に以下のように明記されています。

こちらは、時々誤った認識で「予め予定されている検査入院であってもSAE報告が必要だ」と教えてくる先輩がいますので、注意しましょう(経験談)!
そして、 SAEが生じた際の対応については、GCP上では以下の通り規定されていますね。
(第48条 治験中の副作用等報告)
2 治験依頼者が治験を依頼する場合にあっては、治験責任医師は、治験使用薬の副作用によると疑われる死亡その他の重篤な有害事象の発生を認めたときは、直ちに実施医療機関の長に報告するとともに、治験依頼者に通知しなければならない。この場合において、治験依頼者、実施医療機関の長又は治験審査委員会等から更に必要な情報の提供を求められたときは、当該治験責任医師はこれに応じなければならない。
そのため、SAEが生じた時には、治験責任医師から依頼者と実施医療機関の長宛に「重篤な有害事象等に関する報告書」を提出してSAEがあったことを通知しなければいけないことが分かりますね。
重篤な有害事象が発生したら、治験責任医師→治験依頼者&実施医療機関の長に通知しなければならない(報告書を提出しなければならない)。
SAE発生後の全体的なフロー
まずは、SAE発生後にどのように対応が進むかの全体像を見ていきましょう。詳細は後述しますので、新人の方は「なんとなくこんな感じかぁ~」くらいでOKです。
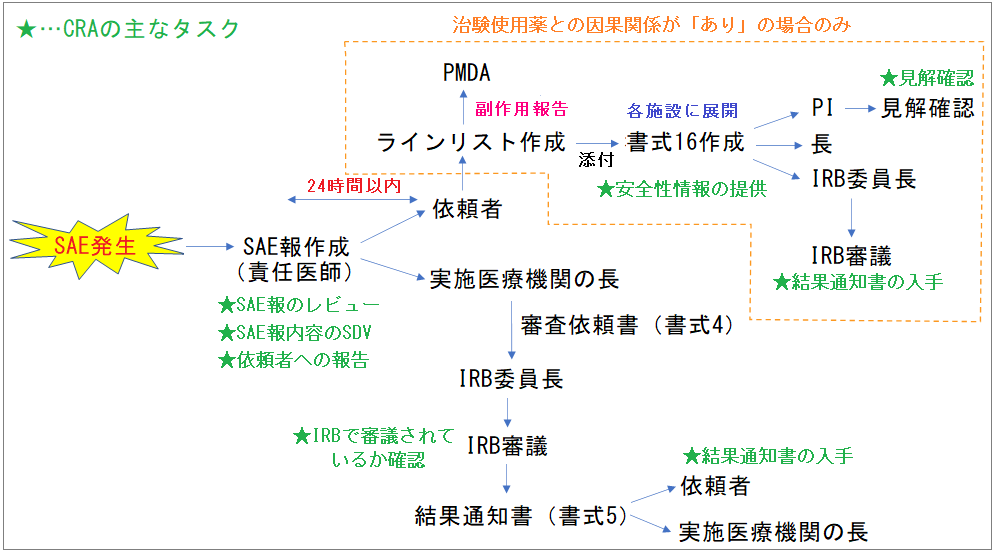
SAEが発生して施設から報告書が提出されると、一般的には上記のようなフローでの対応となります。
私が後輩にSAE発生時の対応を教える時には、大まかに3つのセクションがあることを伝えています。
②SAE報の内容のDA/SDV
③SAE報のIRB審議の確認
まずは、この3つを押さえておいて詳細を詰めていけば良いと思っています。
色々な対応が発生するため、新人のうちはてんやわんやしてしまうかもしれませんが、1つ1つのフローをしっかりと確実にこなしていくことでしっかりと慣れてきます。
新人の方は、暗記というよりは「流れを理解すること」に重点を置いてみましょう。
SAE発生時に登場する資料
SAE対応をする中では色々な資料が登場します。ここでは1つ1つについてじっくり説明をしていきます。
重篤な有害事象に関する報告書(書式12、14、19)
SAEが発生してまず始めに登場する資料です。
医薬品の場合は書式12、医療機器の場合は書式14、再生医療等製品の場合は書式19が重篤な有害事象に関する報告書(不具合含む)に該当します。
実務では「SAE報」と呼ばれることが多々あるので、「SAE報」という呼び方にも慣れておきましょう。
SAE報は更に「速報」と「詳細報」に分岐していますので、その関係性についても理解しておく必要があります。

以下は医薬品のSAE報(速報)になります。
20180710.png)
この書式は、かつて「書式12-1」と呼ばれていたものなのですが、いわばSAE報の表紙にあたるような位置付けの資料になります。
SAEが発生した直後は詳細がまだ分からないため、取り急ぎ、この書式12を治験責任医師がSAEを知り得てから24時間以内に提出することが規定されているプロトコルが大半を占めています。
詳細は付いていない上の形式で報告された報告書を業界では「速報」と呼んでいますので、この呼び方にも慣れておきましょう。
また、最近では統一書式への押印省略がほとんどですが、一部の資料のみ押印が必要な資料がある場合があって書式12も治験責任医師の押印が必要な施設も中にはあったりします。
なので、自分の担当が押印ありの施設かそれとも押印無しの施設かはしっかりと事前に確認しておく必要があります。


そして、これらの資料は詳細報になりますが、詳細報は試験によって提出期限がまちまちなのでしっかりとプロトコールで確認をしておく必要があります。
ちなみに、私の会社では「速やかに」という表現に留めるようにしていてキツキツの期限設定はしていないのですが、多くの場合は「速報を提出してから○日以内」などの制限を付けているかと思います。
今回の記事のテーマは、この詳細報の書き方・レビューの着眼点についてがメインとなりますが、まずは資料紹介をしていきたいので次の資料の説明に移ります。
●SAE報の連絡方法(E-mailの宛先は?EDCでの報告?)
●PIの押印は?(必要の施設と不要の施設があるので確認)
●治験継続の可否は?
●PRT、ICF改定の要否は?
●SAE報提出後のSDVの決まりは?(プロジェクトによって違う)
●詳細報の期限はどのくらいか?
治験審査依頼書(書式4)

SAEが発生したらSAE報が作成されますが、治験責任医師から実施医療機関の長がSAE報を受け取ったらIRBで審議にかけなければいけません。
なので、実施医療機関の長→IRB委員長宛に「治験審査依頼書」が作成されます。
しかし、依頼者が入手すべき資料ではないため慣れないうちは存在を忘れがちですが、必須文書SDVなどでしっかりと確認するようにしておきましょう。
●IRBまでの間に第1報、第2報、第3報など、複数発生している場合は漏れに注意!!
治験審査結果、指示・決定通知書(書式5)

IRB審議が終われば結果通知書(書式5)が作成され、CRAが入手・確認することになります。
既に提出されているSAE報が漏れなくIRB審議にかかっていて承認が貰えていることを確認していきます。
SAE報が出てからIRB審議がされ、その結果が通知されまでの一連の流れをしっかりとイメージしておけば確認資料の見落としリスクを下げることが出来ますので、この流れは完璧にしておきましょう。
書式5は委員出欠リストでミスがあることも…なので、最後まで油断をしないで確認することが必要です(おや、この出欠リスト…)。
安全性情報等に関する報告書(書式16)

報告されたSAEが、治験使用薬との因果関係があった場合(つまり副作用の場合)は、SAE報を受け取った依頼者はPMDAに報告をします。
ここでポイントなのが、副作用の場合ということです。
副作用ではない場合は、PMDAへの報告は原則されませんので、書式16によって他の施設への展開はされません。
SAE報作成時・確認時のポイント

CRAとしてはもちろんのこと、依頼者の立場からもSAE報は相当な数を見てきました。
そこで、受領するSAE報でよくあるミスやSAE報作成時に留意しておいてほしいポイントをまとめてみました。
そもそもSAE報はなぜ作成するか
とても重要なのことなのですが、忘れられがちなので改めて書かせていただきます。
SAE報は、生じたSAEについて治験使用薬の安全性をなるべく正確に評価をするために必要な情報が報告された資料です。
もう少し具体的に書くと、「このSAEはそもそもなぜ生じたのか?」、「治験薬の安全性に問題はないのか?」、そのあたりをしっかりと評価をしていくための重要な判断材料になるということです。
安全性情報は市販後にも収集されますが、市販後の場合は発生件数が分かっても分母が分からない(全体で何錠投与されたか分からない)ので正確な発生頻度を出すことは出来ません。
一方で治験は、母集団が少ないということはありますが、選択除外基準でしっかりとバックグラウンドが揃えられており、かつ治験薬投与例数も分かるので正確な発生頻度を算出することが出来ます。
CRA(特にCRCさん)は、あまり触れないかもしれませんが治験の安全性情報はDSURに集約され、世界的に情報共有もされます。
また、PMDAへの副作用報告の際は、報告医の判断(治験の場合はPIですね)とは別にその報告を見て依頼者としての判断も併せて報告することになります。
ですので、SAE報の内容でミスリードが起きてしまうと依頼者判断が誤ってしまうリスクが上がってしまうことになります。
それだけ、治験で収集される安全性情報は重要な安全性評価材料となることを最初に改めて知ってもらいたいです(上記のことは、依頼者でないと分からないことも多いと思いますので…)。
SAE報確認時の個人的なおすすめ
SAE報確認時のやり方は人それぞれだと思いますが、私がおすすめする確認方法は以下の通りです。
まずは、詳細記載用様式の3枚目の「経緯」と「コメント」を確認します。

「経緯」と「コメント」には、医師の判断を含めSAEについての概要が記載されていますので、その内容とSAE報の1~2枚目(場合によっては4枚目)の情報に齟齬が無いかを確認していきます。
例えば、今回の例の被験者さんの場合は治験薬投与後に実施した軟性膀胱鏡検査を行ったことが原因で尿路感染症となり、抗生物質等で処置がされています。
経過には、「発熱」、「嘔吐」、「WBCとCRPの上昇」が記載されており、別事象として報告されていないことから、これらはSAE「尿路感染症」の随伴症状と判断されていることが分かりますので、尿路感染症の転帰を確認する際にはそれらの随伴症状の状況にも目を配っておく必要がありますね。
このように概ね頭の中でイメージをしてから内容を読み進めていくとSAE報のレビューの精度が上がってきます。
●SAE「尿路感染症」の治療薬として「セフトリアキソンNa」や「レボフロキサシン錠」が処方されている。
●SAE発現の原因は、軟性膀胱検査に伴うものであると判断されている。
●「発熱」と「嘔吐」は回復、「CRP上昇」は改善傾向であると判断されている。
その他、SAE報のレビューをする際には、「今回のSAE報で直してほしいという項目」と「次回のSAE報で直してほしいという項目」にしっかりと分けて考えることが重要だと思っています。
何でもかんでも「すぐ直して下さい」は施設の負担を増してしまうだけなので、Criticalではない修正については「次回のSAE報作成時に修正をして下さい」とする方が良いでしょう。
新人のうちは、Criticalかどうか分からないこともあると思うので、その辺りはしっかりと先輩に確認を取るようにしましょう。
「詳細情報の有無」、「未知既知」、「転帰」
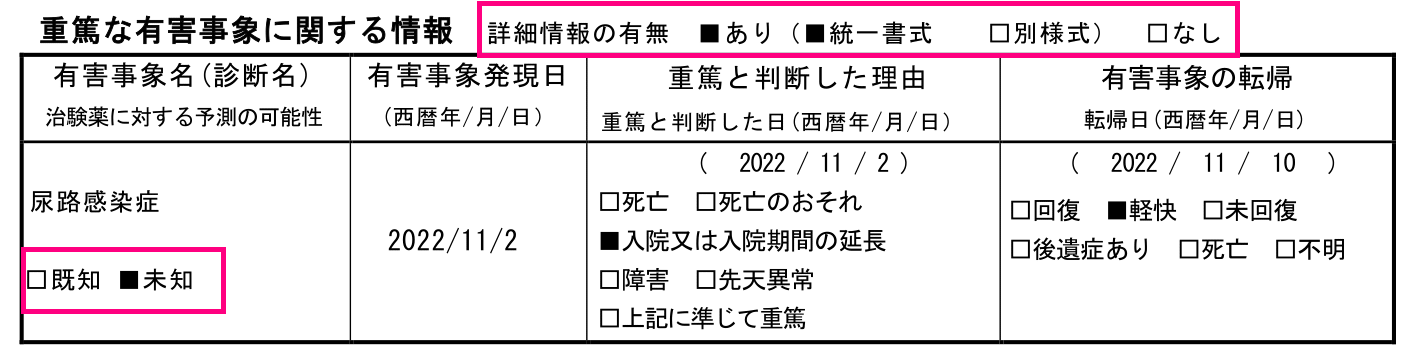
「詳細報の有無」にチェックが付いていなかったり、未知既知の選択が誤っていたりという部分は過去に何回か指摘したことがあります。
未知既知については、一般的に治験薬概要書(IB)に載っているかどうかで判断をするかと思いますが、SAE報を急いで作成していると見落としてしまうこともあるかと思います。
ですので、目視で確認せずに「Ctr+F」で事象名を検索をかけるなど電子的に探してみる方が良いかと思います。
また、試験によってはIBの電子版の提供が不可という依頼者もあるかと思うので、その場合は面倒でしたがExcelで未知既知リストを作成して施設に参考資料として渡すなどをしていました。
転帰については、今回は「改善傾向」と医師判断がありますので「軽快」で良いのですが、例えば転帰が「回復」となっていたらコメントとの齟齬が出てきます。
その際は、コメントの「改善傾向(つまり「軽快」)」と1枚目に記載されている「回復」のどちらが正しいのかを確認する必要が出てきますね。
SAEに関連すると思われる合併症や処置等

こちらの項目には重篤な有害事象に関連すると思われる情報を載せます。
今回のSAE「尿路感染症」は、軟性膀胱鏡検査を実施したことによって生じたものであると判断されていますので、「高血圧症」や「腰部脊柱管狭窄症」は「重篤な有害事象に関連すると思われる情報」には該当しません。
ですので、こちらの項目には記載すべき内容ではないことが分かります。
また、「膀胱癌」が既往歴として載っていますが、ここは少し微妙なラインかなと思います。
なぜなら、軟性膀胱鏡検査が膀胱癌切除後のフォローアップ検査として実施しているものである場合は、間接的に「重篤な有害事象に関連すると思われる情報」にあたる可能性があるためです。
個人的には記載不要だと考えますが、記載してあっても指摘する程ではないかもしれませんね(ここは意見が分かられるかもしれません)。
ちなみに、「高血圧症」や「腰部脊柱管狭窄症」は確かに関係無いけど、別に載せてても良いのでは?と思う方もいるかもしれませんが、個人的にはやはり載せるべきではないと思います。
なぜなら、「そもそもSAE報はなぜ作成するか」でお話をした通り、SAE報の目的は「このSAEはそもそもなぜ生じたのか?」をしっかりと評価するための材料となるためです。
その判断をするための情報に関係の無い余計な情報が混ざっていたら…それは本来あるべき姿ではないですよね?
「高血圧症」や「腰部脊柱管狭窄症」を入れていても指摘をしないReviewerも多いと思いますが(大体の理由は施設にわざわざ指摘を出したくないため)、将来この薬を使う患者さんにとってそれは医薬品開発のあり方として本当に正しいと思いますか?
その辺りを自問して考えてみるのも良いと思います。
重篤な有害事象発現時に使用していた薬剤
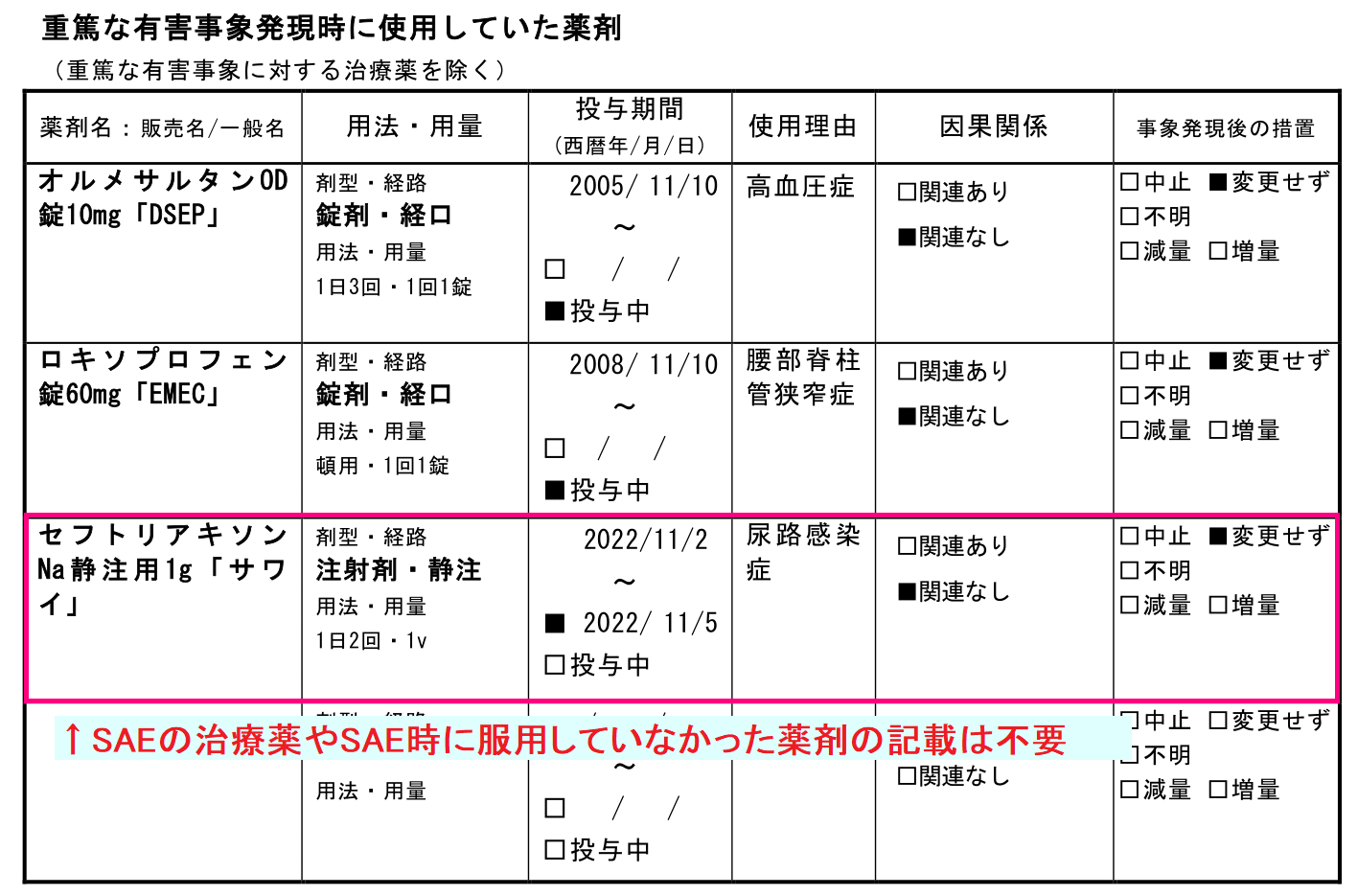
こちらも「SAEに関連すると思われる合併症や処置等」の考え方と同じですね。
重要なのは、「重篤な有害事象発現時に使用していたこと」と「重篤な有害事象に対する治療薬を除くこと」です。
なぜなら、上記以外の「SAE発現時に使用していなかった薬剤」と「SAEに対する治療薬」は物理的にSAE発現の原因にはならないためです。
大切なのでもう1度書きますが、SAE報は「このSAEはそもそもなぜ生じたのか?」を評価するための重要な材料です。
ですので、明らかに関係がない薬剤についてはやはり載せるべきではないでしょう。
今回の例では、私の場合は、「セフトリアキソンNaはSAEに対する治療薬ですので、次回は削除をお願いできますでしょうか」というFBをします。
重篤な有害事象発現に関連すると思われる臨床検査結果
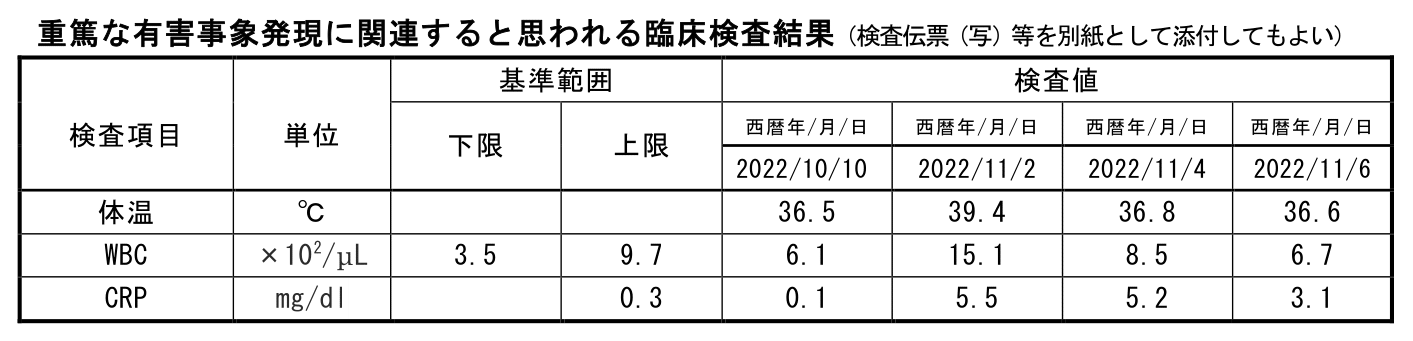
臨床検査値などが絡む有害事象の転帰判断は、「検査値が正常値に戻る」or「その被験者のベースラインまで戻る」場合が回復、そこに至らなくても回復傾向にある場合が軽快などが判断基準になってきます。
今回の例では、尿路感染症によってCRPとWBCが上昇しており2022/11/10には正常範囲には入っていないもののピーク時よりも改善傾向にあるため転帰が「軽快」であることに不自然さはありません。
ただ、仮にここで転帰が「回復」となっていた場合には、「なぜ回復と判断されているのか」をしっかりと確認する必要があります。
まとめると以下のような図となります。
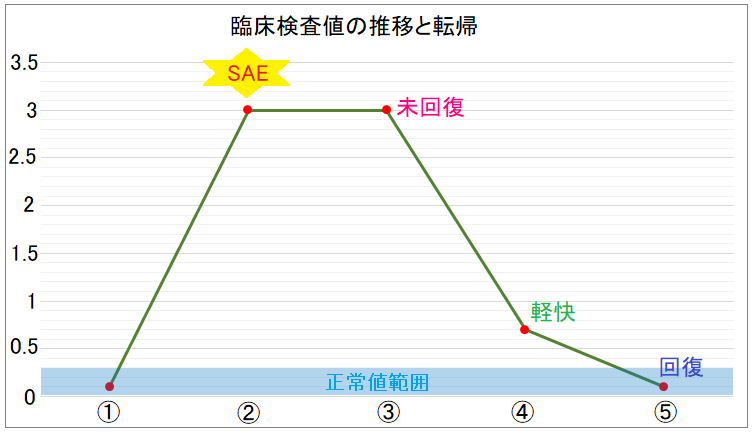
これは、もちろんSAEだけではなくAEについても同じ考え方なので、基本形としてこのようなイメージを持って臨床検査値を見ていけば転帰の違和感には気が付くかと思います。
ここでは臨床検査値について触れていますが、「倦怠感」や「嘔気」など臨床検査値以外の随伴症状がある場合もあります。
そのような場合は、それらの状況も踏まえて総合的に判断されているかどうかという視点で確認していくことが重要です。
まとめ
今回はSAE報についてまとめていきました。
SAEについては、研修でも重点的に教わる部分かと思いますが、GCP上の文言だけでイメージをしようとしてもなかなか難しいものです。
今回は実例を出しながらどのような観点で考えてレビューをしていけば良いのかを示しましたので、新人のみなさまにとって少しでもSAE理解の助けになれば嬉しいです。
SAEが起きると何かと慌ただしいですが、慣れてしまえば対応は流れるように出来ると思いますのでそれまで頑張っていきましょう!
がついに始動!.jpg)




