近年注目を浴びているMuse細胞ですが、2023年2月14日の三菱ケミカルグループ(4188)のニュースリリースにより、同細胞を用いた再生医療等製品の開発の中止が発表されました。
しかし、開発の中止のきっかけの1つとなった治験の結果について製剤効果が過小評価されているのではないかという疑義が浮上しています。
そこで、今回の件について製薬メーカーの臨床開発職の視点からなるべく分かりやすく考察をまとめていきたいと思います。
 のりす
のりす 治験業界でかれこれ10年以上働いています。Twitterもやっています。今回も業界の方以外でも分かりやすいように解説をしていきます
Muse細胞とは
 出典:株式会社カネカ公式HP
出典:株式会社カネカ公式HP
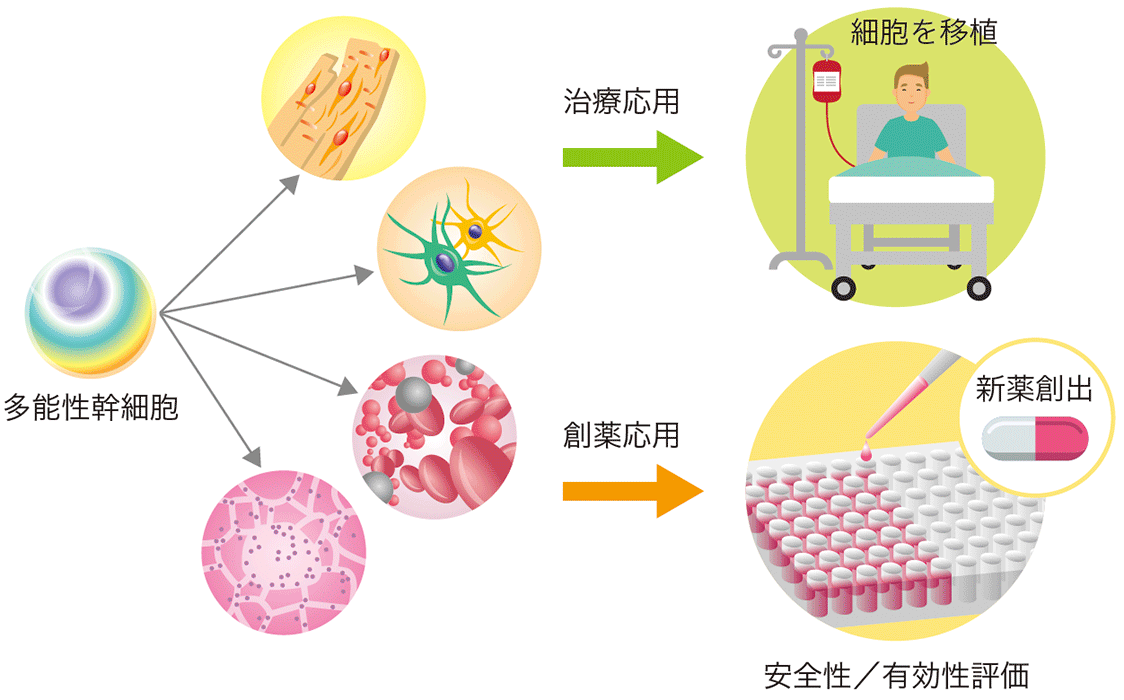
Muse細胞は、ES細胞やiPS細胞に続く「第3の多能性幹細胞」として2010年に東北大学の出澤真理教授らにより発見されました。
多能性幹細胞は、体を構成する全ての組織細胞を作ることができるため、筋萎縮性側索硬化症(ALS)や脊髄損傷などの治療や創薬にも応用できることが期待されています。
期待度が高い一方で課題もあり、ヒトiPS/ES細胞から分化誘導をして移植用細胞を作製する過程で腫瘍化リスクの危険性があることが再生医療にとっての障壁として立ちはだかっていました。
しかし、Muse細胞は分化誘導を必要とせずに生体内に投与することで自発的に分化するという特徴があるため、腫瘍化リスクが低い再生医療として注目を集めているわけですね。
このように近年注目を浴びているMuse細胞ですが、2015年からは、三菱ケミカルグループの生命科学インスティテュート(LSII)により開発コードCL2020として臨床開発が進んでいました。
| 対象疾患名 | 開発相 | 臨床試験情報 |
| ST上昇型急性心筋梗塞 | Ⅰ/Ⅱ | JapicCTI-183834 |
| ST上昇型急性心筋梗塞 | Ⅱ/Ⅲ | JapicCTI-195067 |
| 脳梗塞 | Ⅰ/Ⅱ | JapicCTI-184103 |
| 表皮水疱症 | Ⅰ/Ⅱ | JapicCTI-184563 |
| 脊髄損傷 | Ⅰ/Ⅱ | JapicCTI-194841 |
| 筋萎縮性側索硬化症 | Ⅰ/Ⅱ | jRCT2063200047 |
| 新型コロナウイルス(SARS-CoV-2)感染症に伴う急性呼吸窮迫症候群 | Ⅰ/Ⅱ | jRCT2043210005 |
| 対象疾患名 | 開発相 |
| ST上昇型急性心筋梗塞 | Ⅰ/Ⅱ |
| ST上昇型急性心筋梗塞 | Ⅱ/Ⅲ |
| 脳梗塞 | Ⅰ/Ⅱ |
| 表皮水疱症 | Ⅰ/Ⅱ |
| 脊髄損傷 | Ⅰ/Ⅱ |
| 筋萎縮性側索硬化症 | Ⅰ/Ⅱ |
| 新型コロナウイルス(SARS-CoV-2)感染症に伴う急性呼吸窮迫症候群 | Ⅰ/Ⅱ |
今回の騒動の状況整理

2023年2月14日にMuse細胞の発見者である東北大学の出澤教授により記者会見が開かれました。
記者会見には、治験調整医師や治験に参加をした施設の医師も同席しており、心筋梗塞の治験の結果として提示されたデータに大きな乖離があり、製剤の効果が過小評価されたのではないかという指摘がなされました。
緊急記者会見は午後2時から開催されましたが、その緊急記者会見に先立ち、同日付で三菱ケミカルのニュースリリース「Muse細胞を用いた再生医療等製品(CL2020)の開発中止について」にてMuse細胞を用いた再生医療等製品の開発中止が発表され、波紋を広げています。
以下はYoutubeに掲載されている会見の動画になります。
関係者の整理
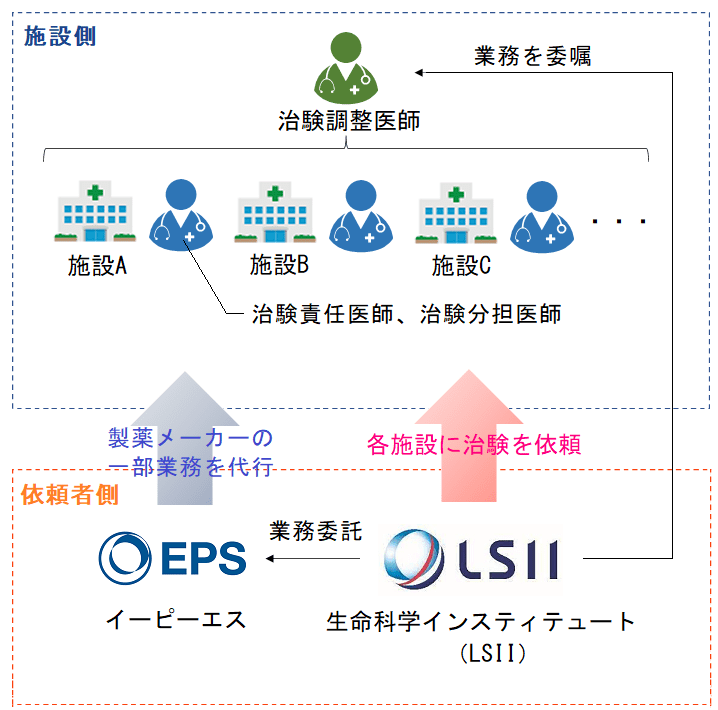
治験での立場は、「施設側(治験を行う施設など)」と「依頼者側(製薬メーカーなど)」に大きく二分されます。
また、治験をおこなうためには多くのリソースが必要になりますので、製薬メーカーはその業務の一部を委託することが多く、今回は生命科学インスティテュート(LSII)がEPSに業務を委託していたという構図になります。
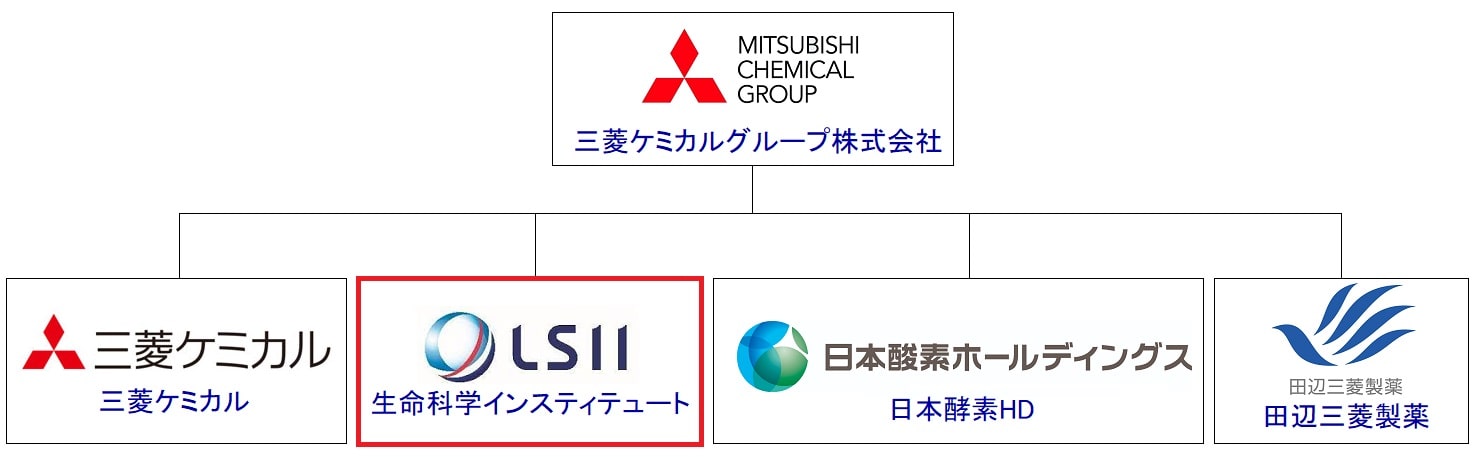
そして、今回の治験を担当していた生命科学インスティテュートは、三菱ケミカルグループのうちの1つになるため、Muse細胞を用いた再生医療等製品の開発中止は三菱ケミカルグループからニュースリリースで発表があったということですね。
問題点の整理
今回の記者会見で主な問題点として指摘されていたことは「施設側のデータと依頼者側のデータの乖離」です。
今回の治験では、施設側で撮影された画像は依頼者側に提出され、依頼者側によって解析が行われる運用でした。
そして、依頼者側による解析の結果出た結論は、プラセボ群と比較して実薬群は有意差を示さなかった…つまり、効果が認められなかったということですね。
依頼者側からこの結果を受け、治験で提出した被験者のデータを施設側で解析したところ、依頼者側が提示してきたデータとの乖離があり、何故なんだ…ということになっています。
ちなみに、施設側で解析した結果ではポジティブな結果が出ていることから、依頼者側は「今回の治験について有効性を過小評価しているのでは?」という疑義が出ています。
この問題を解決するために、施設側は依頼者側に対して解析データの開示を求めていますが、依頼者側は開示を拒否しているという状況のようです。
●解析データの開示について依頼者側は拒否をしている。
厚生労働大臣のコメント
2023年2月17日に開かれた加藤厚生労働大臣の会見で記者より本件についての見解を大臣に問うやり取りがありました。
以下やり取りの抜粋になります。
記者:
三菱ケミカルが多能性幹細胞ミューズ細胞を使った再生医療製剤の開発中止を14日に発表しました。再生医療の推進は政府の重要政策ですが、開発中止をどう受け止めていますか。また、今後新たな開発パートナー探しが難航した場合、ミューズ細胞のこれまでの知見や技術が国外へ流出されることも懸念されますが、この点をどうお考えですか。大臣:
三菱ケミカルグループの生命科学インスティテュート社がミューズ細胞を用いた再生医療等製品を開発中止する旨発表されたことは承知しています。個別事案でありますので具体的なことは差し控えたいと思いますが、再生医療の推進は非常に重要だと考えております。その上で今後再生医療の実用化に向けて様々な相談等がある場合にはしっかりと対応していきたいと考えています。
記者:
もう1点、同日14日にミューズ細胞製剤を使った治験を担当した医師、また発見者である東北大の教授が記者会見し、三菱ケミカルがまとめた治験結果報告書に対して疑義があり、データを開示して病院側の記録と照合するよう求めました。厚生労働省として三菱ケミカル側に何らかの指導等を行う考えはおありでしょうか。大臣:
報道で、都内で緊急会見が開かれ同社に対してデータの照合・確認をその方々が求められていることは承知しております。ただこれは当事者間の個別の案件でありますから、政府としては発言を差し控えさせていただきたいと思います。
あくまで今回の件については、当事者間の個別の案件であり政府としてのコメントは差し控えるというものでした。
今後の展開次第ではありますが、「治験での結果はネガティブであった」という現状を踏まえると、現段階ではMuse細胞製剤の新たな開発パートナー探しは難航する可能性があるかと思います。
逆に、例えば「今回の治験での解析方法等に問題があった」という結果が得られたのであれば、開発パートナーが見つかる可能性も高まるのではないかと推察しています(私がプロジェクトを引き受ける際にはそのような観点を実際に確認すると思います)。
臨床開発職の視点からの考察

今回の記者会見の内容から、一体何が起こっているのかを一臨床開発職として考察していきたいと思います。
色々と深い部分まで考察をしているのですが、なるべく分かりやすく説明できるように意識していきます。
治験を意図的に失敗させる企業側のメリットを感じない
最初に感じたのは、仮に治験の結果を過小評価していたとして、「なぜわざと治験を失敗させようとするのか」という点でした。
今回は心筋梗塞の治験でのお話でしたが、三菱ケミカルグループのプレスリリースによると、「Muse細胞を用いた再生医療等製品CL2020の開発を中止」とあるため、複数走っているパイプラインの開発が中止されることになっているかと思います。
依頼者はどこを見ている?CROを選定する時の基準をまとめてみたの記事ではCROに委託をした場合の費用の紹介をしましたが、CROに委託するだけでも1試験あたり数億かかりますし、医薬品開発にはこれ以外のコストも非常にかかります。
そのため、一般的に考えて三菱ケミカルグループ側(EPSも同様)が治験を意図的に失敗させるメリットは皆無に等しく、ここに大きな違和感を覚えました。
つまり、「意図的に治験を失敗させた」とは考えにくく、あったとしても「運営上のミスによりデータが誤って処理された」なのではないかと思っています。
中央画像判定機関(Central reading center)との結果の齟齬
では、どうして実際に施設側のデータと依頼者側のデータで乖離が起きているのかということですが、個人的には中央画像判定機関が大きく影響しているのではないかと推察しています。
動画の内容から今回の治験では以下のようなフローで治験のデータが収集され、解析されていたと考えられます(動画では「EPSのデータベース」と言われていましたが、もしかしたらマイクロンが用意したサーバーでは?と思ったりも…)。
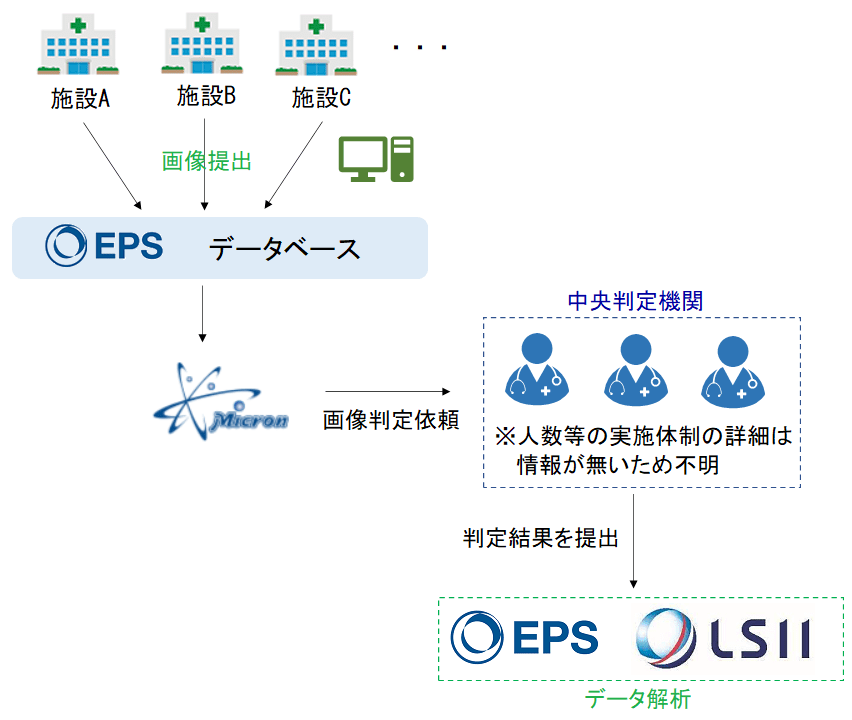
各施設の電子カルテに保存された生データ(画像)を治験用にデータベースにアップロードし、その画像を中央判定機関で読影した後、EPSとLSIIにより統計解析がされたという流れですかね。
今回の治験では、画像からインジェクション・フラクション(EF、左室駆出率)を求める訳ですが、評価者により評価のバラつきがある可能性もあり、各施設の評価と中央判定機関での評価に乖離があることまでは想定の範囲内だと考えます。
ただ、今回はその乖離の程度が大きいことが問題になっているわけですので、中央判定機関関連で乖離が大きくなる可能性を考察していきます。
中央判定委員が適切に選出されていない可能性
今回はCROであるマイクロンが中央判定委員(動画では「第三者」と言われていますね)を選出して読影を依頼していますが、まずは「適切な医師に依頼をしていたか」という点は知りたいですね。
当然のことながら、中央判定委員は施設から提出された画像からEFを出すわけですから専門的な知識を有している必要があります。
中央判定委員としてお願いをする際には、併せて中央判定委員としての適格性のチェックも行っているはずですので、チェックリスト等を確認すれば状況確認ができます。
仮にここで適切な委員の選出ができていなかったとすると、適切にEFが出せていないわけですので、その後の解析の結果もおかしなことになりますよね?
中央判定委員がデータを意図的に過小評価した可能性
中央判定委員は、キーオープン(被験者が実薬群かプラセボ群かの開示)をする前に、つまり盲検化で画像を読影しています。
ですので、中央判定委員が読影を依頼された画像の被験者が実薬群なのか、それともプラセボ群なのかは分かりませんので中央判定委員が故意に実薬群に不利になるような読影をすることは不可能です。
実は本来想定している以上にEFの評価のバラつきが大きい可能性
「EFの評価のバラつきは少ない、あったとしても僅か」という前提のもとでしたが、そもそも実はEFの評価のバラつきは評価者によって大きく異なるのであれば、治験の運用が完璧であったとしても今回のような施設側と依頼者側での結果の乖離が生まれることになります。
私は循環器の専門ではないので詳しくは分からないのですが、少し調べてみたり何人かからお話を聞いた感じですとEFは評価者によってバイアスが入ることもあるということでしたので、もしかしたらこの可能性もそれなりにあるかもしれませんね。
プログラムに不具合があった可能性は考えられる
被験者の割付情報(実薬群かプラセボ群かの情報)と中央判定機関から提出されたデータは別々の外部データになっていて、最終的には紐付けが行われますが、その紐付けに不具合があった可能性は考えられます。
その他、統計解析をする際にはSASプログラムを組んでいますが、コーディングに不備があった場合などには本来と異なるデータが出力される可能性は考えられます。
ただ、EPSは大手CROですのでその辺りの初歩的なミスはしないのではなかなとは思いますがね…
また、上記よりも確率はぐっと下がりますが、被験者の割付情報のシステムに不備があった場合も同じく結果に影響を及ぼすはずです。
施設での治験薬投与のミスの可能性
今回の治験は二重盲検試験という種類の治験ですので、施設の医師も製薬メーカー側も被験者に実薬が投与されているのかプラセボが投与されているのか分からない状態でやっています。
被験者の登録をすると、その被験者に投与する治験薬の治験薬番号が発出され、その番号の治験薬を投与することになりますが、誤った番号の治験薬を投与していた場合、結果に影響を及ぼす可能性があります。
つまり、以下のような感じですね。
【治験薬の情報】
No.1:実薬
No.2:プラセボ
【シチュエーション】
被験者AにNo.1の治験薬を投与するようシステムからの指示があったが、間違えてNo.2の治験薬を投与した場合、システム上では被験者Aは「実薬群」として記録されるが、実際にはプラセボが投与されていることになっている。
ただ、この場合No.2を投与するようシステムから指示が出た時に「No.2の治験薬が無い」ということになるのでまず気が付くことでしょう。
その他、治験ではモニタリングというものが実施されています。
実際には、治験薬が投与された際には治験薬管理表という資料が施設で作成されますので誤った治験薬が投与されていた際にはこのモニタリングでも察知できるはずです。
上記から施設での治験薬投与ミスの可能性はかなり低いものと考えています。
そもそもの解析方法が良くない可能性は低い
治験を開始する前には、対面助言でPMDAと試験計画の認識を擦り合わせていることが一般的です。
もし認識を擦り合わせないで治験を進めた場合、薬事承認申請をする段階で「その評価方法では認められない」ということにもなりかねませんので、そのようなことが無いように製薬メーカーはPMDAに試験デザインについて相談をするわけですね。
ですので、「そもそもの解析方法がダメだった」ということはあまり考えにくいかと思っています。
ただし、もし試験デザインについてPMDAと詰めていなかった場合は解析方法に問題がある可能性も残されることになります。
今後の展開について
今回の施設側の要望について、私個人の目線からどのように見えているのかを少しばかりお話をしていきたいと思います。
もちろん、オープンになっている情報からの判断になりますので、実際にオープンになっていない情報を知れば見解が変わることもあるという前提のもとお話をしていきます。
偽薬と実薬の情報の開示
施設側は、偽薬と実薬の入力が正しいものであることをドクター達にも納得のいく形で確認し、それを提示して欲しいという要望をしています。
これを示すには、システムで自動的に割り振られた各被験者の治験薬番号とキーオープンによって開示された治験薬番号の情報(偽薬か実薬かの情報)、そして被験者個々のEFの数値をまとめた表を三菱ケミカル側が提示することになるかと思います。
「偽薬か実薬かの入力」は手打ちではなくシステム的に入力されているはずですので、そこに「入力ミス」という人為的なミスの可能性は考えにくいです。
有り得るとしたら偽薬か実薬か割り振るシステムのアルゴリズムの不具合ということになりますが、アルゴリズムに不具合が無いかの説明はもはやIT分野になりますので果たしてIT分野以外の方が理解できるのかどうかという点も気になります。
そしてこの要望の厄介な点は、「ドクター達にも納得のいく形で」というやや抽象的な表現が使用されていることです。
具体例が示されていないため、「どのように説明してもドクター達からの納得が得られない」という自体になるのではないかと懸念しています。
私がもし依頼者側でこの件を説明するとしたら、システム的な不具合が発生していないことを確認した後にシステムによって割り振られた帳票と統計解析で使用された被験者の割付情報の帳票を同時に提出し、その2つの情報に齟齬が無いことを提示し、「被験者毎の割付情報は統計解析時も適切であった」ということを示すと思います。
治験データの再測定
施設側としては、中央判定機関の読影によってEFがどのような数値になっているかを開示し、施設側の数値と大きな乖離があった場合には更に第三者に読影を委託するか、あるいは各ドクターが独自に第三者を指定して数値を出すことを要望しています。
これは、先ほども少し触れましたが治験で定められた手順(例えば、中央判定委員の適格性など)に著しい不遵守がなかった場合には三菱ケミカル側も要望を飲むことは難しいであろうと思います。
そもそも中央判定機関は評価者のバイアスを排除するために設置されるものですので、施設側との数値を見せてしまえば(具体的に示さずとも「乖離があるという事実を知れば」)、第三者の評価にバイアスが入る可能性があります。
なので、「施設側の数値と大きな乖離があった場合には」という条件ではなく「手順上大きな重大な不遵守が認められた場合には」としなければ再度EFを出して再解析することは難しいのではないかと考えています。
ちなみに「データを開示したら良い」というのは確かに一般的にはそう思うかもしれませんが、治験のデータは開発が失敗したとしても機密情報であることに変わりはありません。
ですので、迂闊にデータ開示することは出来ず相当慎重になる気持ちは同じ臨床開発職として理解できます。
まとめ
今回はMuse細胞製剤の治験での疑義について考察をしてみました。
今回の件の真相はオープンになっている情報だけからは推測の域でしかなく、実際のところどうなのかは分かりません。
ただ、私自身も臨床開発職として仕事をしている身として、科学的に有用である医薬品についてはしっかりと評価されて患者様に届いて欲しいと思っています。
治験のルールに則って適切に評価されたデータなのか、今後の展開を注視したいところですね。